↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
De novo peptide sequencing is a critical task in proteomics, aiming to identify amino acid sequences from mass spectrometry data. However, existing methods often struggle with data biases, particularly the under-representation of amino acids with post-translational modifications (PTMs). These biases lead to inaccurate sequencing results, hindering progress in understanding protein functions and disease mechanisms.
AdaNovo is a novel framework that addresses these limitations by calculating the Conditional Mutual Information (CMI) between mass spectra and amino acids. This approach helps to identify amino acids with PTMs by highlighting the importance of their mass shifts in the spectra. AdaNovo also uses a robust training approach based on CMI, improving the models’ accuracy and reliability. Experiments showed that AdaNovo outperforms previous methods, achieving significant improvements in PTM identification and overall sequencing accuracy.
Key Takeaways#
Why does it matter?#
This paper is important because it addresses the critical issue of data bias in de novo peptide sequencing, a crucial task in proteomics. By introducing AdaNovo, it offers a robust and accurate method for peptide sequencing, especially for identifying amino acids with post-translational modifications (PTMs). This significantly advances proteomics research and opens new avenues for studying protein function and disease mechanisms.
Visual Insights#
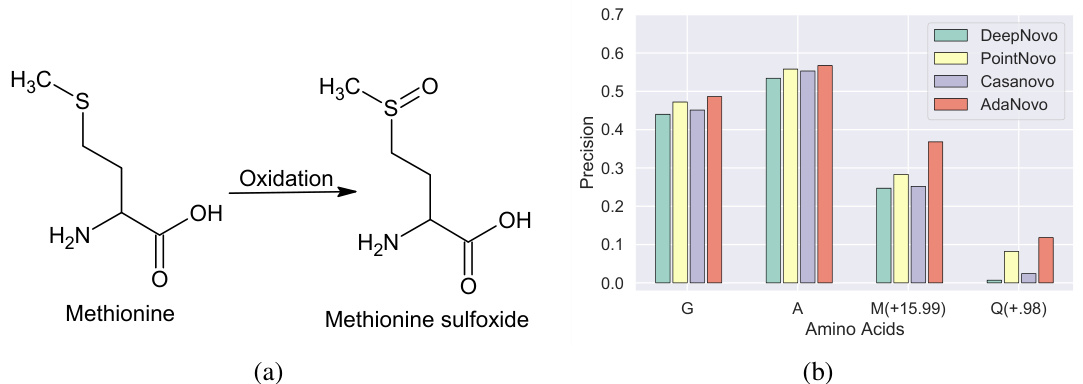
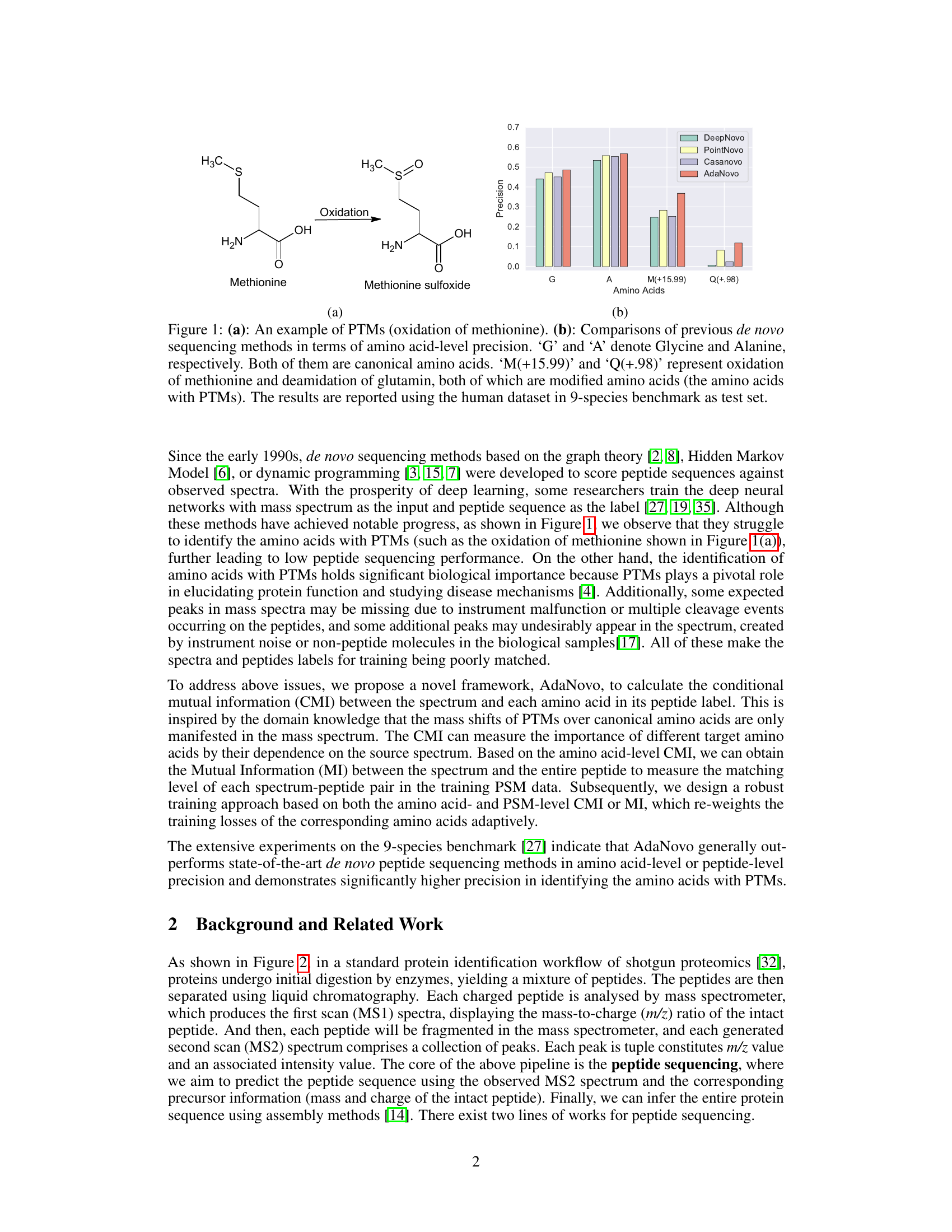
Figure 1(a) shows a chemical illustration of methionine oxidation, a common post-translational modification (PTM). Figure 1(b) presents a bar graph comparing the amino acid-level precision of several de novo peptide sequencing methods. The comparison highlights the superior performance of AdaNovo in identifying both canonical amino acids (Glycine, Alanine) and modified amino acids (oxidized Methionine, deamidated Glutamine), demonstrating its robustness against data biases in proteomics.
This table presents a comparison of the performance of several de novo peptide sequencing models (DeepNovo, PointNovo, Casanovo, and AdaNovo) on a benchmark dataset containing data from nine different species. The table shows the peptide-level and amino acid-level precision for each model on each species, along with average precision scores across all species. The results highlight AdaNovo’s superior performance compared to existing methods.
In-depth insights#
Bias Robustness#
The concept of ‘Bias Robustness’ in the context of de novo peptide sequencing is crucial because training datasets often exhibit biases. AdaNovo directly addresses this by incorporating conditional mutual information (CMI) calculations. This approach weighs training data points based on their reliability and information content. Specifically, AdaNovo focuses on mitigating biases related to the underrepresentation of post-translational modifications (PTMs) and noisy mass spectrometry data. By using CMI, the model learns to distinguish between true signals and noise, enhancing its ability to correctly identify PTMs, a significant challenge for previous de novo methods. The results show that this focus on bias reduction significantly improves the model’s precision in identifying peptides, particularly those with PTMs. This strategy of incorporating domain knowledge to guide the learning process and thus improve robustness against data biases is a significant contribution.
CMI Framework#
A Conditional Mutual Information (CMI) framework offers a robust approach to de novo peptide sequencing by directly addressing data biases in training datasets. The core innovation lies in calculating the CMI between mass spectral peaks and individual amino acids within a peptide sequence. This contrasts with previous methods that predominantly focused on the overall peptide sequence. By leveraging CMI, the framework effectively handles the challenges posed by underrepresented post-translational modifications (PTMs) and noisy spectral data. The CMI calculation provides a weighted measure of importance for each amino acid, allowing the model to learn more effectively from scarce PTM examples. Furthermore, the framework incorporates PSM-level CMI to account for overall spectrum-peptide match quality, improving robustness against the inaccuracies inherent in mass spectrometry. This multi-level approach to CMI-based training empowers a more accurate and reliable de novo peptide sequencing model, significantly improving performance, especially for PTM identification.
PTM Precision#
Analyzing “PTM Precision” requires a nuanced understanding of post-translational modifications (PTMs) and their impact on proteomics research. PTMs significantly alter protein function and are crucial for various biological processes. Accurate identification of PTMs, therefore, is paramount. A key challenge lies in the relative scarcity of PTM-containing peptides in training datasets compared to unmodified sequences. This data bias leads to lower precision in identifying PTMs using standard de novo peptide sequencing methods. Innovative approaches like AdaNovo aim to address this bias by incorporating domain-specific knowledge and using techniques that emphasize PTMs during training and inference. This involves calculating conditional mutual information to robustly handle noisy data and missing peaks in mass spectrometry, resulting in more reliable PTM identification. The effectiveness of such methods is usually evaluated against standard benchmarks to gauge improvements over existing techniques. Further research is needed to explore the limitations of current approaches and develop even more robust methods for PTM identification. Ultimately, improving PTM precision is vital for advancing our understanding of protein function and disease mechanisms.
Adaptive Training#
Adaptive training, in the context of the research paper, seems to address the core challenges of data bias in de novo peptide sequencing. The approach is domain knowledge-inspired, leveraging the inherent properties of mass spectrometry data and its relationship with peptide sequences. This is crucial because existing methods often struggle with variations in amino acid occurrence and noisy spectral data. By employing Conditional Mutual Information (CMI), the model learns to weigh the importance of each amino acid based on its information content in the spectrum, thus effectively mitigating the impact of biased training data. This adaptive re-weighting of training losses, done at both the amino acid and peptide-spectrum match (PSM) level, allows the model to focus on crucial information while downplaying less relevant signals. The result is a more robust and accurate de novo peptide sequencing method, especially effective in identifying post-translational modifications (PTMs), which are often underrepresented in training data.
Future Work#
The ‘Future Work’ section of this research paper on de novo peptide sequencing presents exciting avenues for improvement. Addressing the challenge of identifying previously unseen PTMs is crucial, as current methods struggle with this. Developing more robust training strategies that mitigate the effects of noise and missing peaks in mass spectrometry data is also critical. This could involve exploring advanced data augmentation techniques or incorporating domain-specific knowledge into the model’s architecture. Investigating alternative loss functions beyond the cross-entropy method could improve model accuracy, particularly when dealing with the inherent imbalance in the data caused by infrequent PTMs. Further research on scaling up the model to handle even larger and more complex datasets will be necessary to increase the efficiency of proteomics analysis. Ultimately, integrating AdaNovo with existing database search tools could create a powerful hybrid approach, capable of providing more complete and accurate results. The authors recognize that efficient handling of computational costs is a key consideration for future improvements, as the current model can be computationally expensive. Finally, exploring applications of AdaNovo beyond peptide sequencing and into other proteomics tasks would be valuable.
More visual insights#
More on figures

This figure illustrates the workflow of shotgun proteomics, focusing on peptide sequencing. It starts with biological samples that are enzymatically digested into a mixture of peptides. These peptides are then analyzed using tandem mass spectrometry which produces two types of spectra: MS1 (precursor ion information) and MS2 (fragment ion information). The core challenge is peptide sequencing where the goal is to accurately predict the amino acid sequence of the peptide based on the MS2 spectrum. The figure highlights the MS2 spectrum with signal peaks in blue and noisy peaks in grey, along with the resulting predicted peptide sequence (ATASPPRQK).
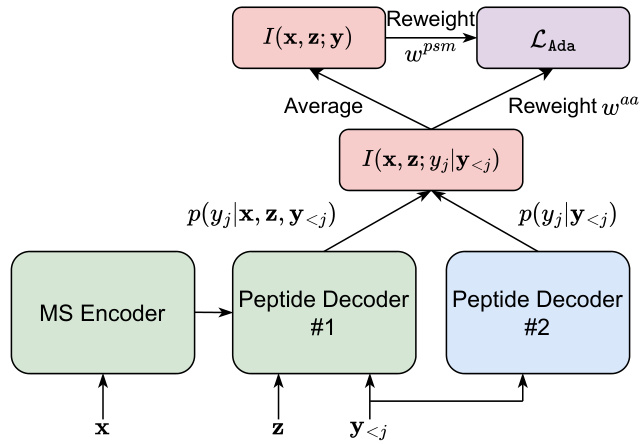
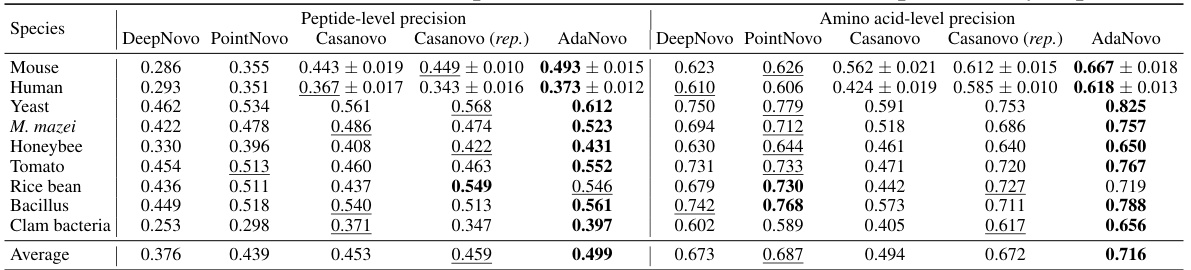
This figure shows the schematic of the AdaNovo framework, illustrating how mass spectrum encoder, two peptide decoders, conditional mutual information (CMI) calculations, and re-weighting strategies work together to improve peptide sequencing. The MS encoder processes mass spectrum peaks (x) and precursor (z) to generate features. Two peptide decoders (#1 and #2) are used; Decoder #1 predicts amino acid probabilities (p(yj|x, z, y<j)), and Decoder #2 predicts probabilities based on the previous amino acids (p(yj|y<j)). The CMI between the spectrum and amino acids is computed (I(x, z; yj|y<j)) and used for re-weighting. PSM-level mutual information is used (I(x,z;y)) for another level of re-weighting, combining to give the adaptive loss (L_Ada).

This figure compares the performance of four different de novo peptide sequencing models (DeepNovo, PointNovo, Casanovo, and AdaNovo) in identifying Post-Translational Modifications (PTMs). The bar chart displays the PTM-level precision for each model across nine different species datasets (Mouse, Human, Yeast, M. mazei, Honeybee, Tomato, Rice bean, Bacillus, and Clam bacteria). AdaNovo consistently outperforms the other models in most species, demonstrating its improved ability to identify PTMs.

This figure shows the effects of hyperparameters s1 and s2 on the performance of AdaNovo model. Hyperparameter s1 controls the effect of amino acid-level adaptive training, and s2 controls the effect of PSM-level adaptive training. The plots show peptide-level and amino acid-level precision for different combinations of s1 and s2 values on the human dataset. The results demonstrate that the optimal values of s1 and s2 significantly influence the model’s performance and that the optimal settings may differ depending on the metric used (peptide-level vs. amino acid-level).
More on tables
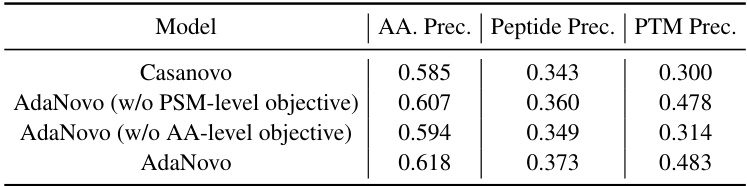
This table presents the ablation study results on the Human dataset, showing the impact of removing either the amino acid-level or the peptide-level training objectives from the AdaNovo model. It compares the performance of the full AdaNovo model against versions without the amino acid-level objective and without the peptide-level objective. The metrics used are amino acid precision (AA. Prec.), peptide precision (Peptide Prec.), and PTM precision (PTM Prec.). The results demonstrate the importance of both training strategies for achieving optimal performance.

This table presents the performance of different models on a mass spectrum dataset with added synthetic noise. The models compared are Casanovo, AdaNovo without the PSM-level objective, AdaNovo without the AA-level objective, and AdaNovo (the full model). The results, specifically amino acid precision (AA. Prec.) and peptide precision (Peptide Prec.), are shown for each model, demonstrating the impact of the noise and the different components of the AdaNovo model on performance. The test dataset used was Clam bacteria.

This table compares AdaNovo with other methods for identifying amino acids with Post-Translational Modifications (PTMs). It shows the amino acid precision, peptide precision, and PTM precision for Casanovo, Casanovo with re-weighting (upsampling), Casanovo with focal loss, AdaNovo without the PSM-level objective, and AdaNovo. The results are based on using the yeast dataset.
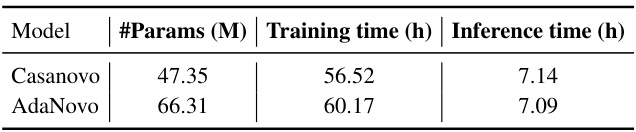
This table compares the computational cost (number of parameters, training time, and inference time) of AdaNovo with Casanovo. AdaNovo, while more accurate, requires slightly more parameters and training time but maintains a similar inference speed to Casanovo.
Full paper#