↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Protein Language Models (PLMs) usually focus on protein sequences, ignoring crucial structural information. This limits their ability to accurately predict protein functions, which are intricately linked to 3D structure. Existing structure-aware PLMs have limitations in representing local structural details and explicitly modeling the relationship between protein sequences and structures.
ProSST solves this by introducing a structure quantization module that converts 3D protein structures into a sequence of discrete tokens, capturing fine-grained structural details. It uses a Transformer architecture with disentangled attention to explicitly model the interplay between protein sequences and structures. Extensive pre-training on millions of protein structures enables ProSST to learn rich contextual representations. The model achieves state-of-the-art results in zero-shot mutation prediction and several downstream tasks, showcasing the effectiveness of its novel approach.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in protein language modeling and structural biology because it presents a novel approach for integrating protein structure information into protein language models. ProSST’s state-of-the-art performance on various tasks and its publicly available codebase will accelerate research in this field, opening new avenues for understanding protein function and developing advanced protein design tools.
Visual Insights#
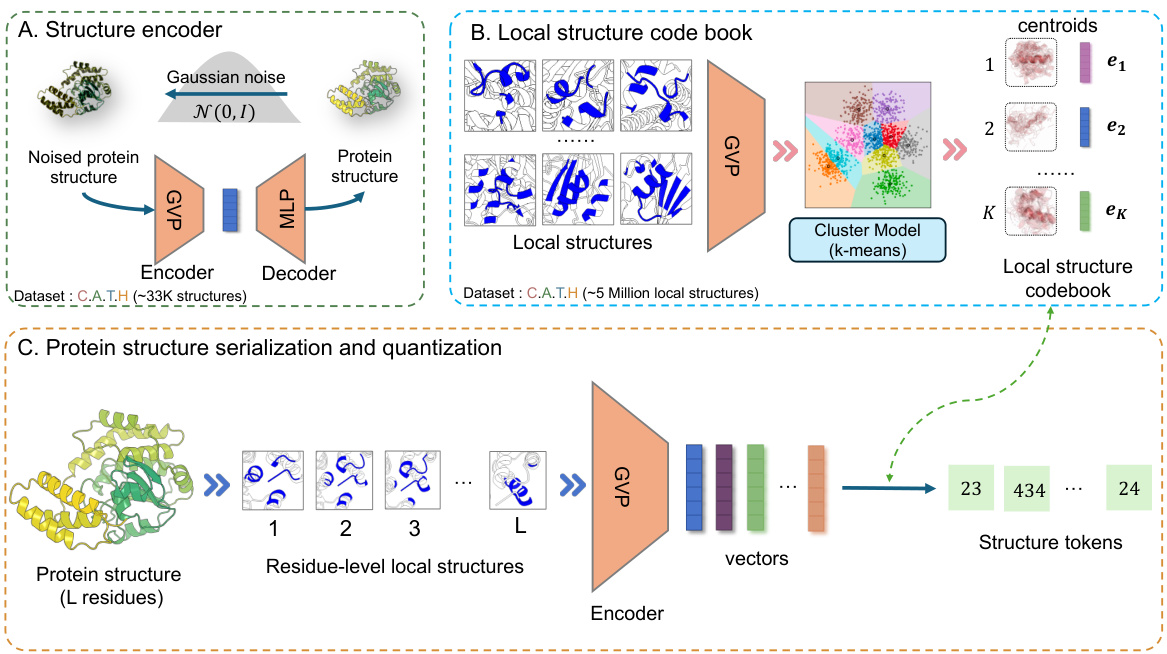
This figure illustrates the three-stage pipeline for protein structure quantization. First, (A) shows the training of a structure encoder using a geometric vector perceptron (GVP) based autoencoder. The encoder takes a protein structure as input, adds gaussian noise, and learns to reconstruct the original protein structure. This trained encoder is then used in the next stage. Second, (B) shows the creation of a local structure codebook. The trained encoder processes a large dataset of protein local structures to produce dense vectors. A clustering model (k-means) is then applied to create a codebook of discrete structure tokens. Finally, (C) shows the protein structure serialization and quantization. The trained encoder processes each residue’s local structure to produce a dense vector, which is then converted to a discrete structure token using the codebook, producing a sequence of structure tokens for the entire protein.
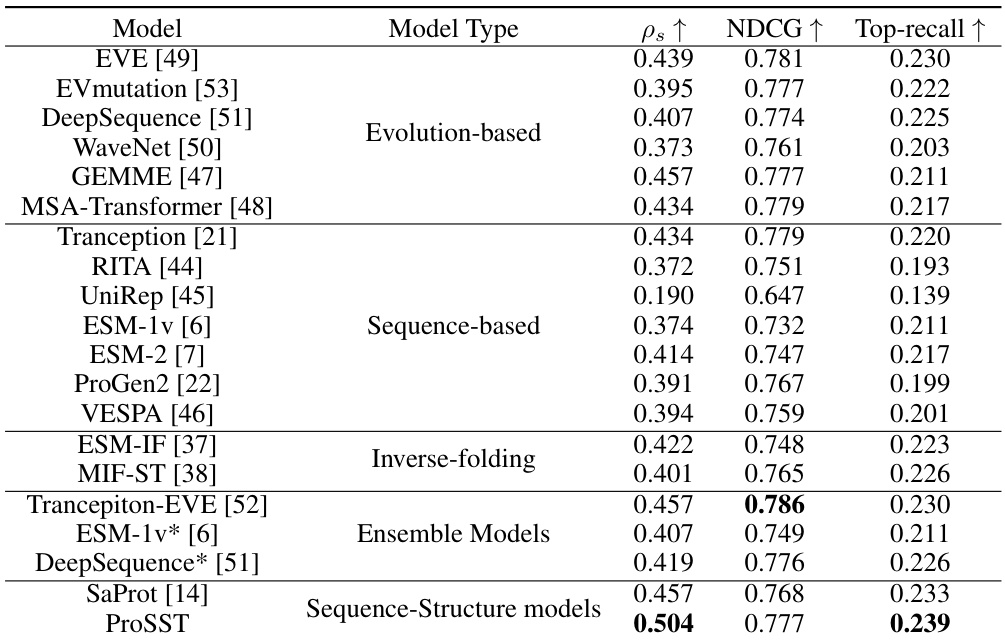
This table compares the performance of ProSST against other state-of-the-art models on the ProteinGYM benchmark for zero-shot mutation effect prediction. It shows the Spearman rank correlation (ρs), normalized discounted cumulative gain (NDCG), and top-recall metrics for each model. The table helps demonstrate ProSST’s superior performance compared to existing methods.
In-depth insights#
Quantized Structure#
The concept of “Quantized Structure” in protein language modeling is crucial for effectively incorporating structural information into model training. Quantization transforms continuous structural data (e.g., 3D coordinates) into discrete tokens, allowing the model to process structural features more efficiently. This discretization simplifies the input, enabling the model to learn relationships between protein sequences and structures more effectively. A key challenge lies in choosing an appropriate quantization method that preserves essential structural information without sacrificing too much detail. Various approaches, such as clustering of local structural representations, or binning based on structural features could be used. The optimal quantization strategy will depend on factors like computational cost, desired level of detail, and the specific downstream task. The success of the approach hinges on developing efficient techniques that effectively capture the critical structural aspects of proteins while maintaining a manageable representation size for the model.
Disentangled Attn#
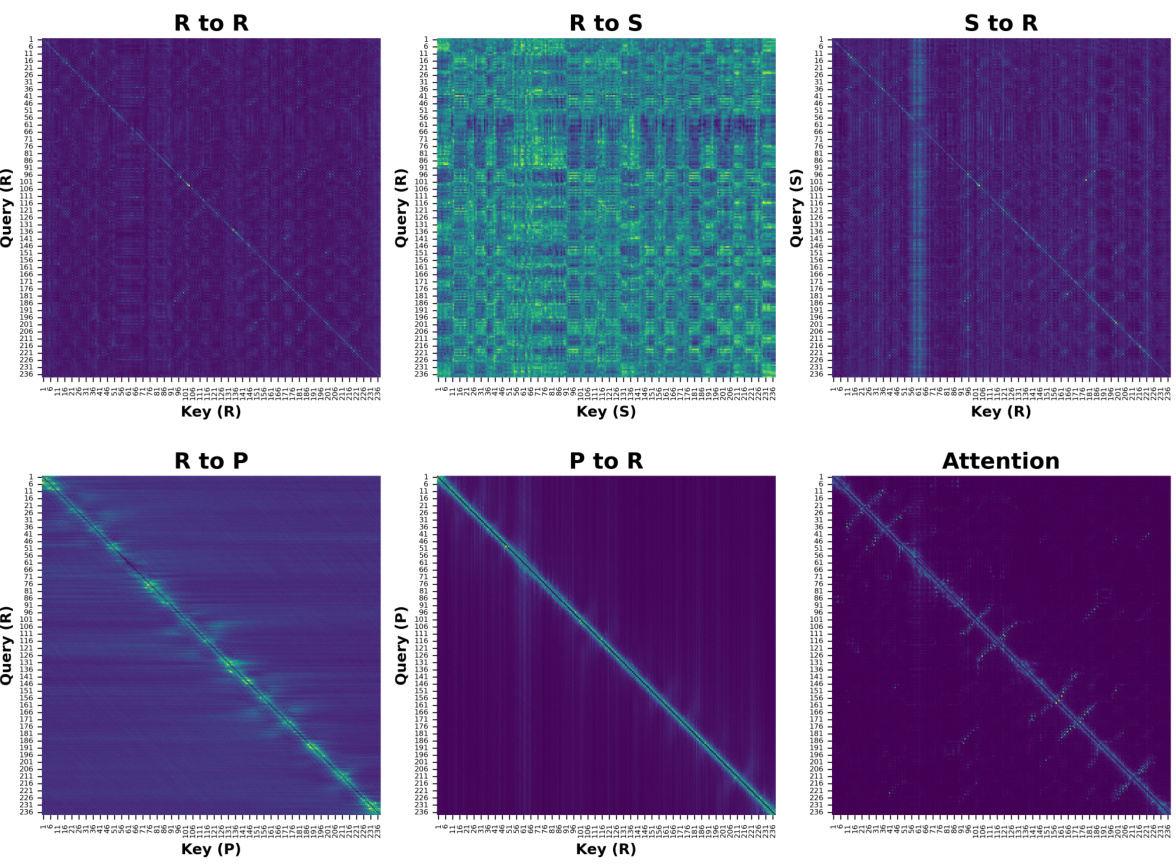
Disentangled attention mechanisms are designed to improve the efficiency and effectiveness of attention in transformers by separating the different aspects of information processing. Instead of a single attention mechanism processing all aspects simultaneously, disentangled attention separates the attention process into multiple independent components, each focusing on a specific type of relationship (e.g., residue-residue, residue-structure, or position-based relationships). This approach enhances the ability of the model to capture complex relationships within the data and reduce the risk of interference or overshadowing between different information channels. The use of disentangled attention in ProSST is particularly valuable because it explicitly models the relationship between protein sequences and structures, enabling the model to integrate both types of information effectively. The effectiveness of disentangled attention in ProSST, relative to traditional self-attention mechanisms, strongly suggests that the decomposition of attention into separate components facilitates a deeper understanding of the contextual relationships. This design improvement allows the model to capture nuances not otherwise evident in traditional approaches.
ProSST Model#
The ProSST model presents a novel approach to protein language modeling by integrating both protein sequences and structures. Structure quantization, using a geometric vector perceptron (GVP) encoder and k-means clustering, translates 3D structures into discrete tokens. This is a significant improvement over existing methods because it leverages a more effective protein structure representation. Disentangled attention allows the model to explicitly learn the relationship between sequence and structure tokens, enhancing the integration of structural information. The use of a masked language model (MLM) objective for pre-training on a large dataset enables ProSST to learn comprehensive contextual representations. Zero-shot prediction capabilities and superior performance on downstream tasks highlight ProSST’s effectiveness and efficiency. The model’s design demonstrates an advanced understanding of protein structure and function, making it a strong contender in the field.
Zero-Shot Mut#
The heading ‘Zero-Shot Mut’ likely refers to a section detailing experiments on zero-shot mutation effect prediction. This implies the model was tested on its ability to predict the impact of mutations on protein function without explicit training on mutation data. This is a significant benchmark as it showcases the model’s capacity to generalize knowledge learned during pre-training to a new task. Success in zero-shot mutation prediction suggests the model has learned robust and comprehensive representations of protein structure and function. A strong performance would indicate a deeper understanding of the relationships between sequence, structure, and function, captured effectively during pre-training. The results in this section would likely be compared to other models and evaluated based on metrics like accuracy and correlation with experimental data. The discussion would likely emphasize the model’s ability to make predictions on unseen data, highlighting its generalizability and potential for applications in drug discovery and protein engineering.
Future Work#
Future work in this area could explore several promising avenues. Improving the efficiency of the structure quantization module is crucial, as it currently presents a computational bottleneck. Investigating alternative quantization techniques, or more efficient encoding methods based on graph neural networks, could substantially enhance performance and scalability. Expanding the dataset to include more diverse protein structures, such as those from less-studied organisms or with complex post-translational modifications, would enable more robust and generalized model training. Furthermore, exploring other downstream tasks, beyond those considered in the paper, will better demonstrate the generalizability and utility of the proposed ProSST model. Investigating the integration of other modalities, such as protein-protein interaction data or experimental binding affinities, would yield richer contextual information and potentially further improve prediction accuracy. Finally, developing a more thorough understanding of the disentangled attention mechanism, and exploring alternative designs that leverage the interaction between protein sequence and structure more effectively would be a valuable research direction.
More visual insights#
More on figures

This figure shows the architecture of ProSST, a Transformer-based protein language model. It highlights the key components: the structure quantization module which converts protein structures into a sequence of discrete tokens, and the disentangled attention mechanism which allows the model to learn relationships between protein residue sequences and structure token sequences. The disentangled attention module is a modified version of self-attention, explicitly modeling interactions between residue tokens, structure tokens, and positional embeddings. This architecture enables the model to effectively integrate structural information with protein sequences for comprehensive contextual representation learning.

This figure illustrates the three stages of the structure quantization module in ProSST. Stage (A) shows the training process for the structure encoder, a neural network that converts 3D protein structures into dense vector representations. Stage (B) depicts the creation of a codebook using k-means clustering on the encoded vectors from a large dataset of protein structures. The codebook maps similar local structural patterns to a discrete token. Finally, Stage (C) demonstrates how a full protein structure is processed, creating a sequence of tokens representing the protein’s structural information.

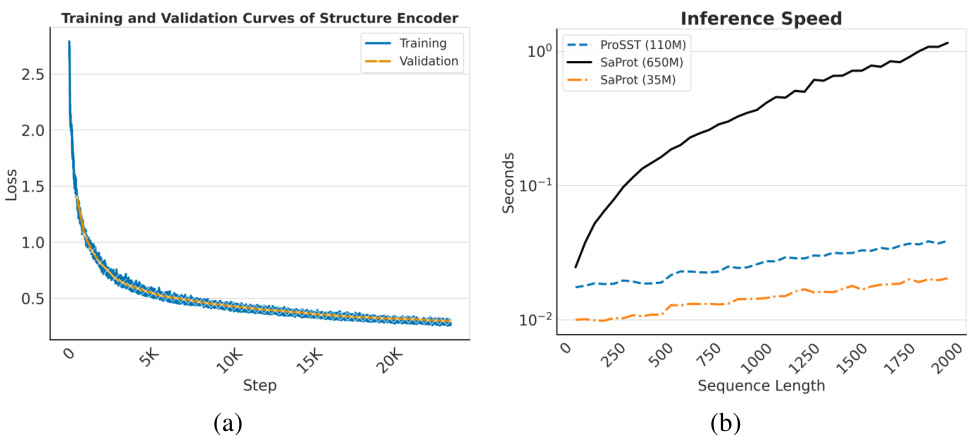
This figure displays perplexity curves, a measure of model performance, for ProSST under various conditions. It shows the effect of ablating different components of the model, including the quantized structure (varying K values) and different components of the disentangled attention mechanism (removing different attention types). The x-axis represents the training step, and the y-axis represents the perplexity. Lower perplexity indicates better performance. The results demonstrate the importance of both quantized structure and disentangled attention for ProSST’s performance.

This figure illustrates the process of converting a protein’s 3D structure into a sequence of discrete tokens. It involves three main steps: (A) Training a structure encoder using a denoising autoencoder to learn a robust representation of local protein structures. This encoder takes a protein structure as input and outputs a dense vector representation. (B) Building a structure codebook by clustering the dense vector representations of millions of local structures from the CATH dataset using k-means. The centroids of the clusters form the codebook, and each centroid represents a discrete structure token. (C) Serializing the protein structure into a sequence of residue-level local structures. Each local structure is then encoded using the trained structure encoder, and the resulting vector is quantized into a discrete structure token using the structure codebook. The final output is a sequence of these discrete tokens representing the protein’s structure.
This figure illustrates the process of converting a protein’s 3D structure into a sequence of discrete tokens. Panel (A) shows the training of a structure encoder, a neural network that learns to represent local structural features as dense vectors. Panel (B) depicts the creation of a codebook by clustering similar local structural vectors into groups, each represented by a unique token. Finally, panel (C) shows how the entire protein structure is processed, creating a sequence of these discrete structure tokens for use as input to the main ProSST model.
The figure shows the architecture of the ProSST model, which is a transformer-based model that uses disentangled attention. Disentangled attention is a mechanism that allows the model to explicitly model the relationship between protein sequence tokens and structure tokens. This enables the model to learn comprehensive contextual representations of proteins, which improves its performance on various downstream tasks. The figure highlights the key components of the ProSST model, including the structure quantization module, the disentangled multi-head attention module, and the feed-forward module. It also shows how the different types of attention are combined to generate the final output.
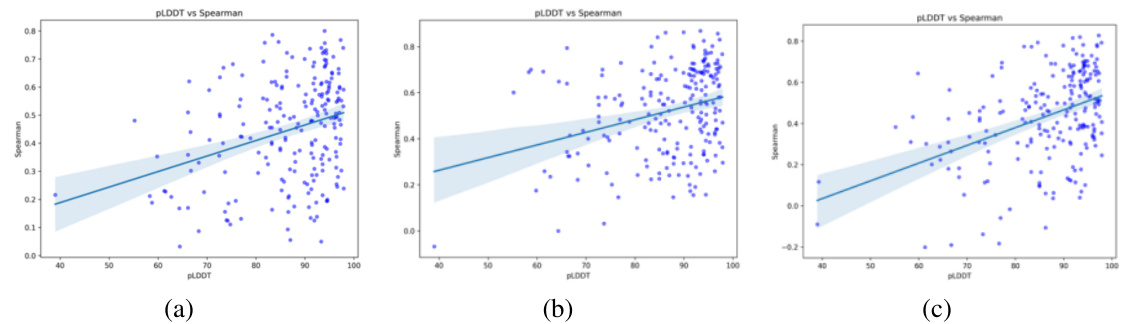
This figure displays the correlation between AlphaFold2’s predicted local structure quality (pLDDT) and the performance (Spearman correlation) of three different protein language models, ProSST, SaProt, and ESM-IF1, on the ProteinGYM benchmark. Each panel shows a scatter plot, with pLDDT values on the x-axis and Spearman correlation values on the y-axis. A linear regression line is also included to visualize the trend. The positive correlation suggests that the accuracy of structure prediction influences the performance of the models. Disordered regions of proteins may affect the prediction accuracy of AlphaFold2, potentially impacting downstream model performance.
More on tables

This table presents the results of supervised fine-tuning experiments on four protein downstream tasks: DeepLoc (protein localization), Metal Ion Binding, Thermostability, and Gene Ontology (GO) annotation prediction (GO-MF, GO-BP, GO-CC). The model ProSST is compared against several baselines (ESM-2, ESM-1b, MIF-ST, GearNet, SaProt-35M, SaProt-650M, ESM-GearNet) across these tasks. The performance metrics used are accuracy (Acc%) for DeepLoc and Metal Ion Binding, Spearman’s rank correlation (ρs) for Thermostability, and maximum F1-score (F1-Max) for GO annotation prediction. The number of parameters (# Params) for each model is also provided. The table highlights ProSST’s superior performance across all tasks, despite having fewer parameters than many of the baselines. Error bars (standard error) are included for ProSST’s results.

This table presents the ablation study on the quantized structure module of ProSST. It shows the performance of ProSST with different numbers of centroids (K) in the k-means clustering model used for structure quantization. It also compares the performance of ProSST using the proposed quantization method with those using Foldseek and DSSP.
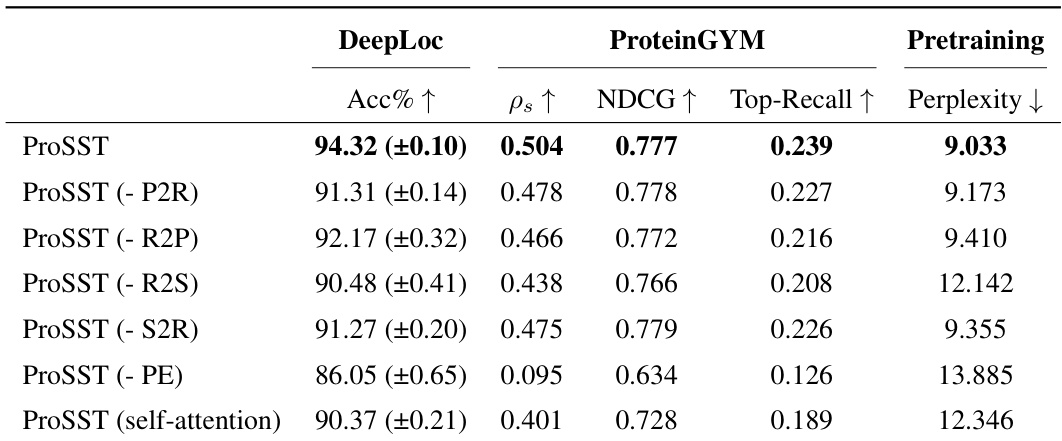
This table presents the ablation study results on the disentangled attention mechanism of the ProSST model. It shows the performance of ProSST model variants with different components removed from the disentangled attention, including residue-to-residue, residue-to-position, residue-to-structure, structure-to-residue, position-to-residue, and positional encoding. The results are evaluated on DeepLoc (Accuracy), ProteinGYM (Spearman’s rank correlation, NDCG, Top-Recall), and pre-training (Perplexity).

This table presents the results of supervised fine-tuning experiments on four protein downstream tasks: DeepLoc, Metal Ion Binding, Thermostability, and GO annotations prediction (MF, BP, CC). It compares the performance of ProSST against several other models, including ESM-2, ESM-1b, MIF-ST, GearNet, SaProt (two sizes), and ESM-GearNet. The evaluation metrics used are Accuracy (%) for DeepLoc and Metal Ion Binding, Spearman’s rank correlation (ρs) for Thermostability, and the maximum F1-score (F1-Max) for GO annotations prediction. The number of parameters (# Params) for each model is also provided.
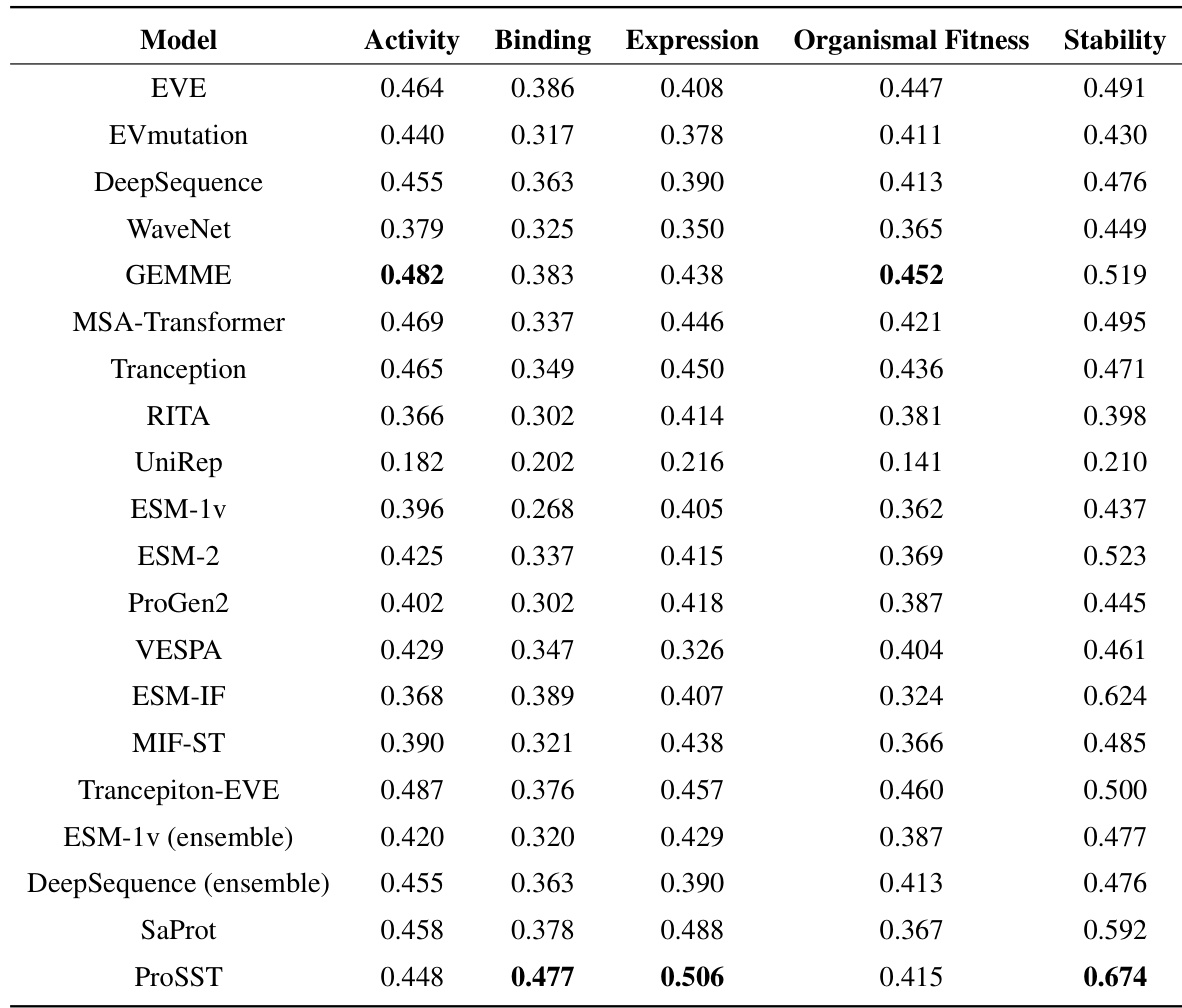
This table compares the performance of ProSST against other state-of-the-art models on the ProteinGYM benchmark for zero-shot mutation effect prediction. It shows the Spearman rank correlation (ps), normalized discounted cumulative gain (NDCG), and top-recall metrics for each model, allowing for a direct comparison of their predictive capabilities.
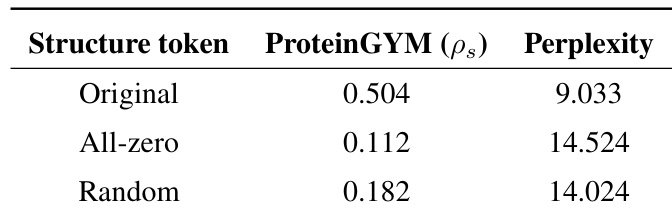
This table presents an ablation study on the impact of structure tokens on the performance of ProSST. It shows the Spearman’s rank correlation on the ProteinGYM benchmark and the model’s perplexity during pre-training using different structure token inputs: the original structure tokens, all-zero tokens, and random tokens. This demonstrates the crucial role of accurate structure information in ProSST’s performance.

This table compares the performance of ProSST models trained with different numbers of centroids (K) in the structure codebook. It shows the accuracy on the DeepLoc task, the Spearman’s rank correlation on the ProteinGYM benchmark, and the perplexity achieved during pre-training. The results demonstrate the impact of the structure quantization module on ProSST’s performance.
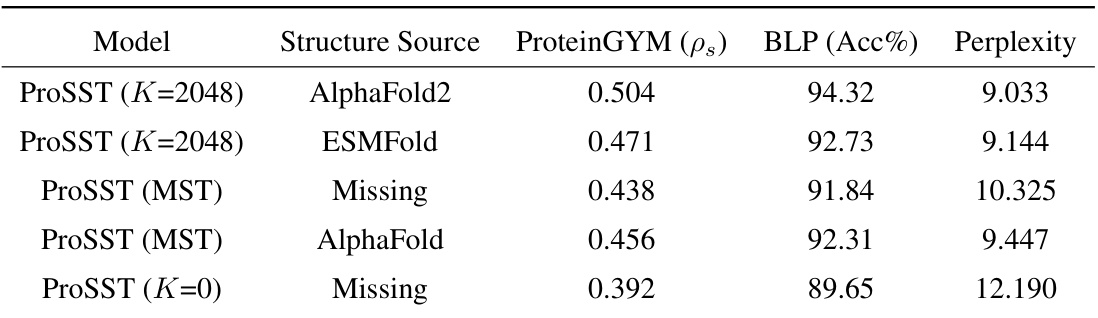
This table presents the results of ablation studies conducted on ProSST using different settings for the number of centroids (K) in the structure quantization module. It compares the performance of ProSST with different structure sources (AlphaFold2 and ESMFold) and training approaches (with and without the structure data) on two downstream tasks: zero-shot mutation prediction on the ProteinGYM benchmark and protein localization prediction (BLP). The results are evaluated using the Spearman rank correlation (ps), accuracy (Acc%), and perplexity. This is intended to illustrate the effectiveness of the proposed structure quantization method and the necessity of structure information for ProSST’s performance.
Full paper#