↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Protein structure prediction heavily relies on Multiple Sequence Alignments (MSAs), which are often incomplete for many proteins, hindering accurate predictions. This research tackles this challenge by developing a novel generative model. Current methods struggle to create MSAs for proteins lacking abundant homologous sequences. This limits the performance of many prediction models.
The researchers introduce MSA-Generator, trained using a unique sequence-to-sequence task, to address the issue of incomplete MSAs. This model successfully generates virtual MSAs that boost the accuracy of existing protein structure prediction models. Extensive experiments show significant improvements in prediction accuracy, particularly for complex proteins, highlighting the potential of generative models in this field.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in protein structure prediction and related fields because it introduces a novel approach to generate high-quality multiple sequence alignments (MSAs). The innovative seqs2seqs pretraining method significantly improves the accuracy of existing protein structure prediction models, especially for proteins lacking sufficient homologous sequences, a major bottleneck in the field. This opens new avenues for improving the accuracy and efficiency of protein structure prediction and related tasks.
Visual Insights#

This figure illustrates the core idea of the paper. The left panel shows the traditional search-based MSA approach, where a lack of homologous sequences for certain proteins results in low-quality MSA and consequently inferior structure prediction. The right panel introduces the proposed MSA-Generator, a generative model that creates high-quality virtual MSAs by enriching existing ones, leading to improved prediction accuracy. The figure visually compares the AlphaFold2 predictions using both search-based and generative-based MSAs, highlighting the superior performance of the latter.

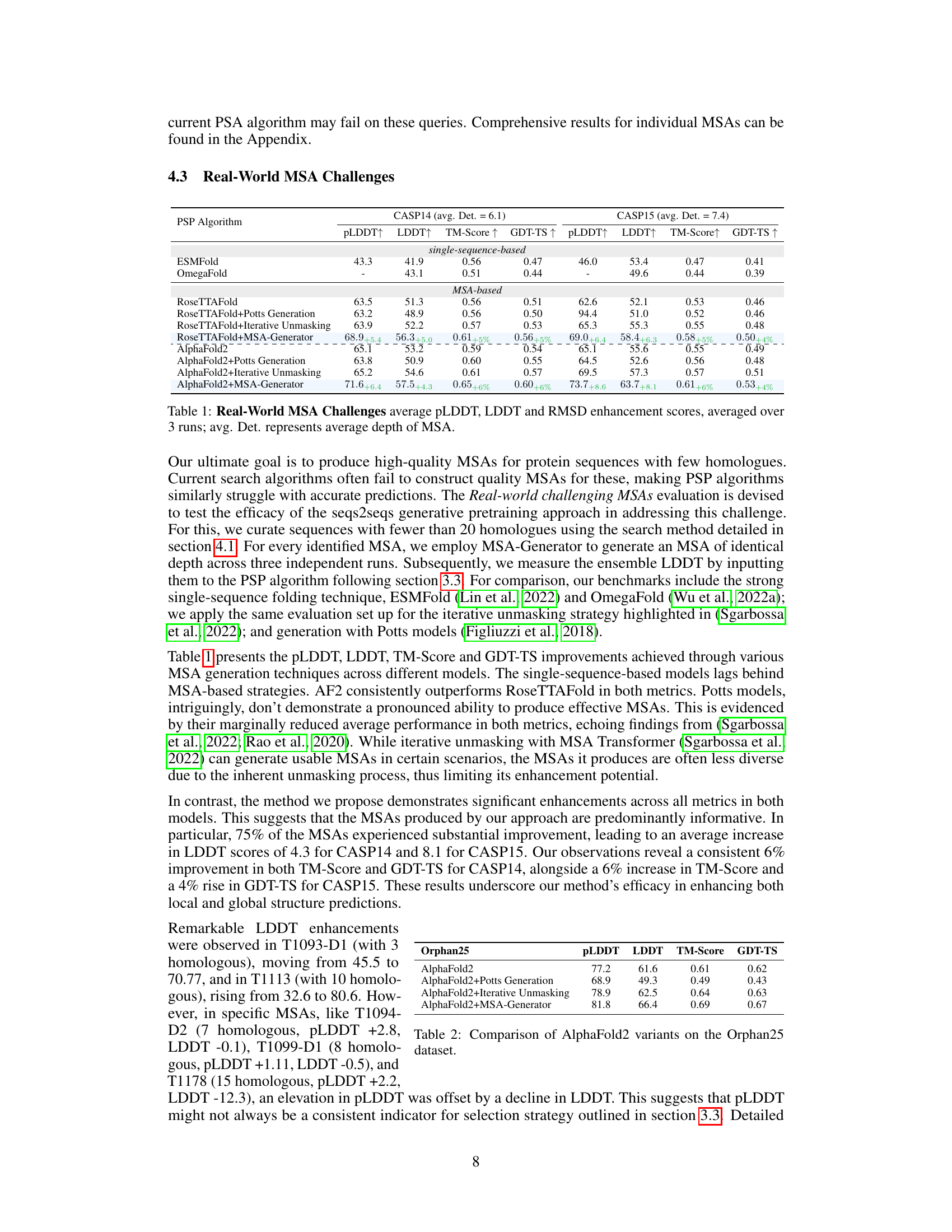
This table presents the results of applying various MSA generation methods to improve the performance of protein structure prediction algorithms (RoseTTAFold and AlphaFold2) on challenging protein sequences from the CASP14 and CASP15 datasets. It compares the performance of these algorithms using standard MSAs against their performance when supplemented with MSAs generated by different methods, including Potts generation, iterative unmasking, and the proposed MSA-Generator. The metrics used to evaluate performance are pLDDT, LDDT, TM-Score, and GDT-TS. The ‘avg. Det.’ column indicates the average depth (number of sequences) in the MSAs.
In-depth insights#
Seqs2seqs Pretraining#
The proposed ‘Seqs2seqs Pretraining’ approach represents a significant advancement in protein structure prediction. By framing the problem as a sequence-to-sequence task, it moves beyond the limitations of traditional methods. Unlike conventional seq2seq models that focus on one-to-one correspondence, seqs2seqs embraces flexibility, enabling the generation of multiple coherent sequences from a single input. This allows the model to capture rich evolutionary information, a crucial factor in predicting protein structures. The use of protein-specific attention mechanisms further enhances the model’s ability to effectively leverage large-scale protein databases. The self-supervised nature of the training process is another key strength, eliminating the need for large labeled datasets, and making the approach more accessible and scalable. The results demonstrate that the model significantly improves the performance of existing structure prediction tools, especially for challenging sequences with limited homologous data. This innovation holds the potential to revolutionize protein structure prediction and open up new avenues of research in related areas of bioinformatics.
MSA Generation#
The heading ‘MSA Generation’ in this context likely refers to methods for creating multiple sequence alignments (MSAs), crucial for accurate protein structure prediction. The paper likely explores novel approaches to generating MSAs, surpassing traditional search-based methods. These new methods could involve using deep learning models, possibly trained on a large protein sequence dataset, learning to predict sequences similar to a given query sequence. This may involve self-supervised learning, using protein-specific attention mechanisms to identify and generate highly accurate and informative MSAs, especially beneficial when dealing with proteins lacking extensive homologous sequences, a common challenge in protein structure prediction. The effectiveness of these generated MSAs would likely be demonstrated by showcasing improved accuracy in subsequent structure prediction tasks. Generative models trained with a sequence-to-sequence approach are likely discussed. The innovation could involve using techniques such as masked language modeling or transformers. Overall, the approach aims to enhance protein structure predictions by addressing a significant bottleneck in traditional MSA generation.
AlphaFold Enhancements#
This research explores AlphaFold enhancements by focusing on the critical role of Multiple Sequence Alignments (MSAs) in protein structure prediction. The core idea revolves around a generative model, MSA-Generator, trained via a novel self-supervised ‘sequences-to-sequences’ task. This approach overcomes the limitation of relying solely on existing, potentially incomplete MSAs, especially for proteins lacking abundant homologous sequences. MSA-Generator produces virtual MSAs that enrich existing ones, resulting in improved prediction accuracy. The study demonstrates significant improvements in LDDT scores when integrating the generated MSAs with AlphaFold2 and RoseTTAFold, particularly for complex proteins. These improvements highlight the effectiveness of the generative approach in boosting the performance of existing state-of-the-art methods. The research also analyzes the characteristics of generated MSAs (diversity and conservation) and rigorously evaluates its efficacy on CASP14/15 benchmarks, showing substantial enhancements. The success of this method suggests a promising direction for future advances in protein structure prediction, extending beyond solely relying on search-based MSA methods. While pLDDT served as the primary selection metric, the authors also examine the direct use of LDDT, highlighting nuances in selecting the most effective MSA for prediction accuracy.
pLDDT Re-evaluation#
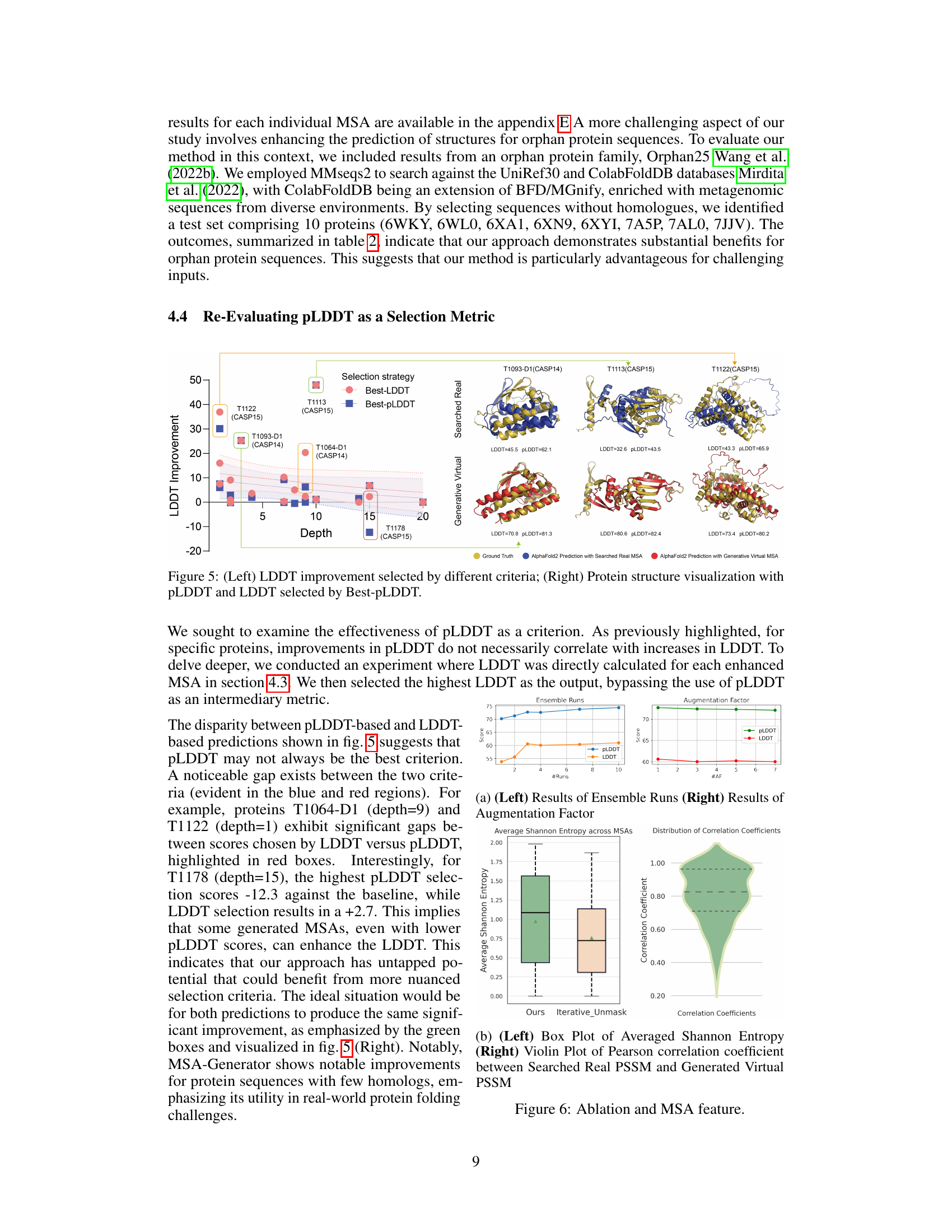
The re-evaluation of pLDDT as a selection metric reveals its limitations as a sole indicator for MSA quality. While pLDDT often correlates with LDDT (Local Distance Difference Test), a key metric for protein structure prediction accuracy, the study highlights instances where high pLDDT scores do not translate to high LDDT, suggesting that pLDDT might not always be a consistent predictor of improved accuracy. This finding underscores the importance of considering alternative selection criteria or a more nuanced approach, possibly involving ensemble methods, to accurately assess MSA quality and select the best MSA for downstream structure prediction tasks. This insight is crucial for optimizing protein structure prediction workflows and demonstrates the need for more sophisticated approaches to evaluating MSA quality and its impact on prediction performance.
Future Directions#
Future research could explore larger-scale models and more extensive training data to further enhance MSA-Generator’s capabilities. Investigating alternative self-supervised tasks beyond seqs2seqs would also be valuable to determine their effectiveness in generating diverse and informative MSAs. Developing more sophisticated evaluation metrics that go beyond LDDT and pLDDT to more accurately assess MSA quality and downstream prediction accuracy is crucial. Furthermore, refining the selection process for the best MSA and exploring efficient strategies for handling computationally expensive large-scale MSAs warrants future investigation. Finally, extending the MSA-Generator framework to other bioinformatics tasks, such as RNA secondary structure prediction or microbial community analysis, could reveal its broader utility and potential impact.
More visual insights#
More on figures
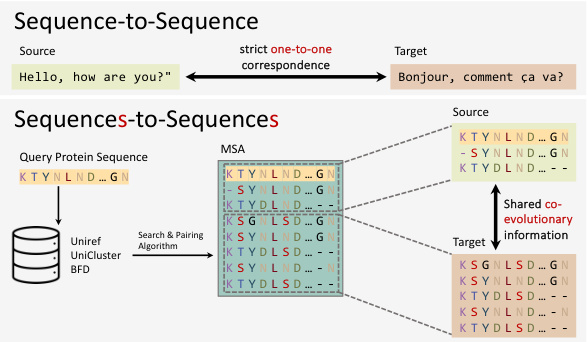
This figure compares the traditional search-based method for Multiple Sequence Alignment (MSA) generation with the proposed generative approach using MSA-Generator. The left side illustrates how limited homologous sequences for certain protein queries result in low-quality MSAs using conventional methods which consequently lead to inferior structure predictions. The right side shows how MSA-Generator produces high-quality virtual MSAs by leveraging large-scale protein databases. These virtual MSAs supplement the existing ones and improve the accuracy of protein structure prediction algorithms.
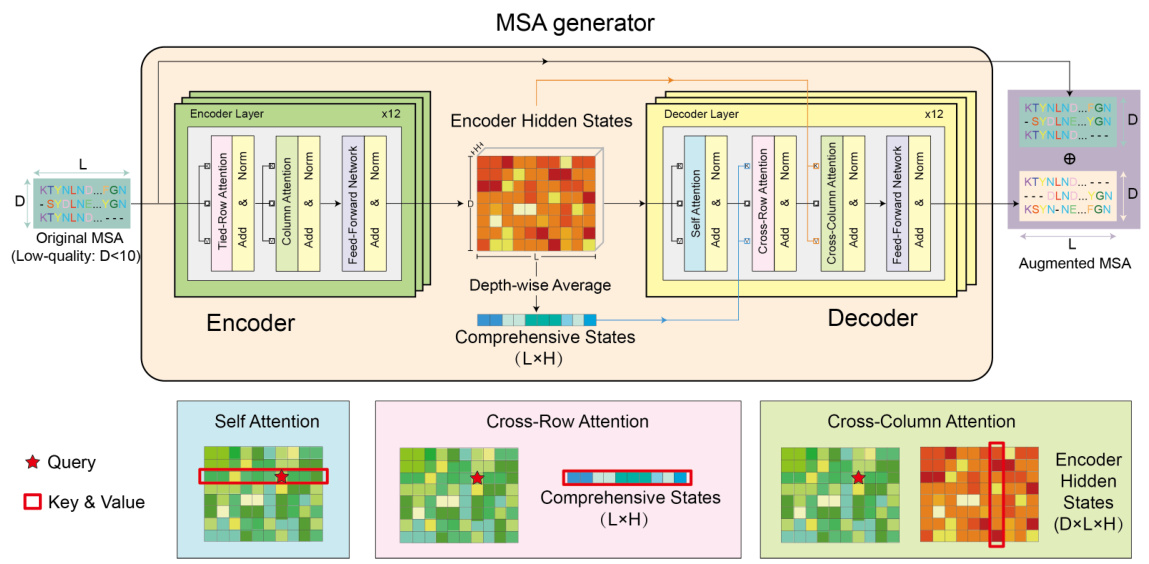
This figure illustrates the architecture of MSA-Generator, a transformer-based model for generating multiple sequence alignments (MSAs). The top part shows the overall architecture, including the encoder, decoder, and attention mechanisms. The bottom part provides a detailed explanation of the attention mechanism used in the model, showing how the model attends to different parts of the input sequence to generate the output. The figure highlights the use of tied-row attention, cross-row attention, and cross-column attention to efficiently capture global structural information from input MSAs.

This figure displays the results of comparing the performance of Alphafold2 with generated virtual MSAs versus real MSAs on artificial challenging protein sequences. Panel (a) shows violin plots comparing the LDDT (Local Distance Difference Test) scores for real MSAs, baseline MSAs (reduced number of sequences), and virtual MSAs generated by MSA-Generator. Panel (b) is a scatter plot showing the correlation between the LDDT of real and virtual MSAs, highlighting the close approximation achieved by the generated MSAs. Panel (c) provides a breakdown of the LDDT improvements in different intervals, comparing both virtual MSAs against baseline and real MSAs. The overall results demonstrate that MSA-Generator produces virtual MSAs comparable to or even surpassing real MSAs in accuracy.
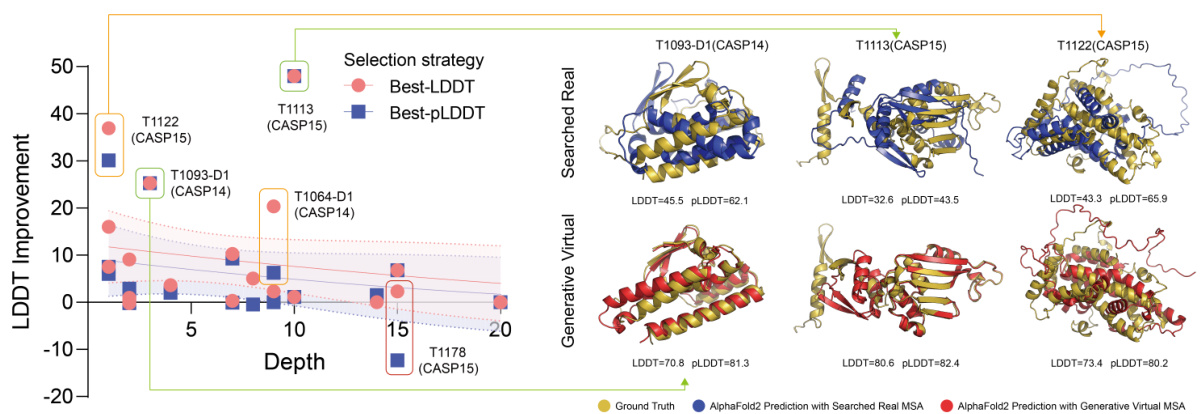
This figure shows a comparison of LDDT improvements achieved using two different selection strategies: Best-LDDT and Best-pLDDT. The left panel is a violin plot showing the distribution of LDDT improvements for several proteins at various MSA depths. The right panel provides 3D structural visualizations for selected proteins (T1093-D1, T1113, T1122, and T1178), comparing the ground truth structure with AlphaFold2 predictions using searched real MSAs and generative virtual MSAs. The figure highlights the differences in prediction accuracy based on the choice of selection metric and demonstrates the impact of MSA-Generator in improving the LDDT scores, especially for proteins with lower MSA depths.
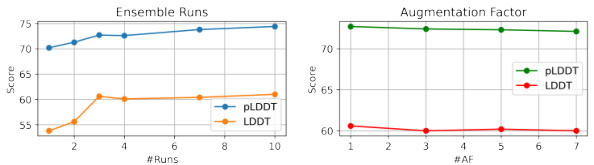
This figure presents the ablation study and MSA feature analysis. The left panel (Ensemble Runs) shows the impact of increasing the number of ensemble runs on both pLDDT and LDDT scores. The right panel (Augmentation Factor) shows the effect of varying the augmentation factor on the same metrics. The results indicate that while more ensemble runs improve performance, the returns diminish beyond a certain point. Similarly, there is limited improvement from increasing the augmentation factor. These results guide the choice of the optimal number of ensemble runs and augmentation factors used in the main experiments.
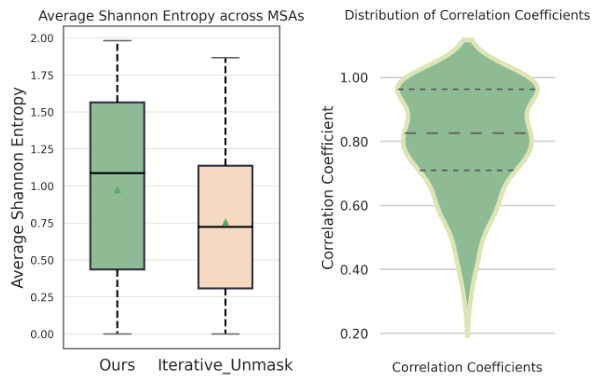
This figure shows two subfigures. The left subfigure is a box plot showing the average Shannon entropy across MSAs for the proposed method (Ours) and the Iterative_Unmask method. The right subfigure is a violin plot showing the distribution of Pearson correlation coefficients between searched real PSSM and generated virtual PSSM. These plots demonstrate the effectiveness of the proposed method in generating diverse and accurate MSAs.

This figure visualizes the Multiple Sequence Alignments (MSAs) generated by MSA-Generator. It shows three examples (T1060s2-D1, T1093-D1, T1096-D1) of MSAs, where different colors represent different amino acids. The top half of each example displays the original MSA, while the bottom half shows the augmented MSA generated by MSA-Generator. This visualization helps to illustrate the diversity and conservation characteristics of the generated MSAs compared to the originals.
More on tables
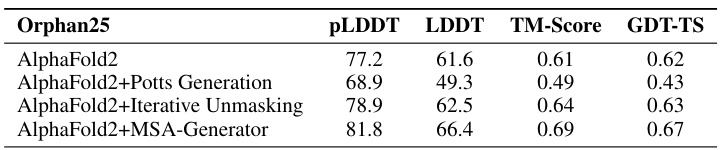
This table presents a comparison of the performance of four different methods on the Orphan25 dataset, a challenging set of protein sequences with limited homologs. The methods compared are AlphaFold2 alone, AlphaFold2 combined with Potts Generation, AlphaFold2 with Iterative Unmasking, and AlphaFold2 enhanced with the MSA-Generator. The metrics used for comparison are pLDDT, LDDT, TM-Score, and GDT-TS, which are standard measures of protein structure prediction accuracy. The results show that the addition of MSA-Generator significantly improves AlphaFold2’s performance on this difficult dataset.
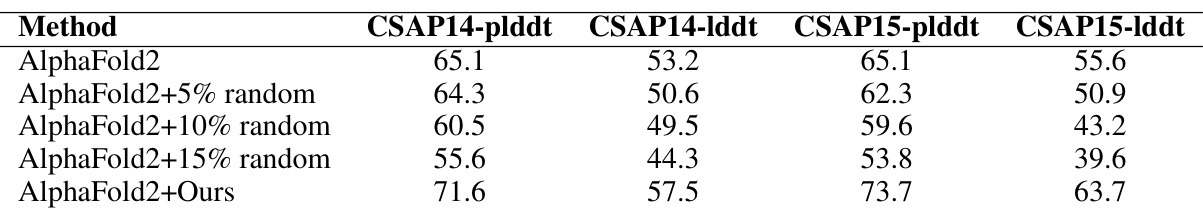
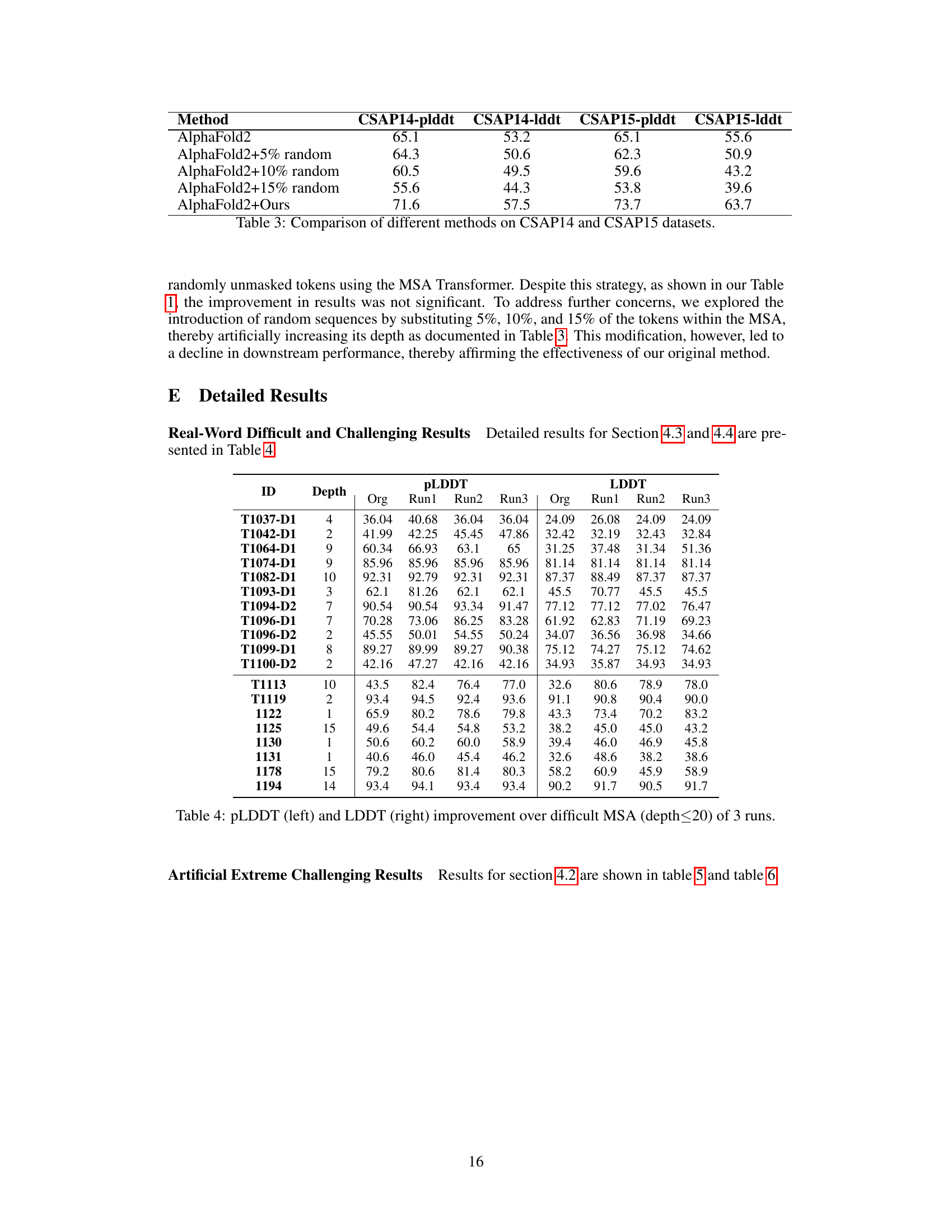
This table compares the performance of AlphaFold2 with and without several modifications on the CASP14 and CASP15 datasets. The modifications include adding different percentages (5%, 10%, 15%) of random sequences to the MSA and using the MSA-Generator method proposed in the paper. The results are presented as average pLDDT and LDDT scores, showing the impact of each modification on the accuracy of protein structure prediction.

This table presents the results of experiments conducted on real-world challenging MSAs. It compares the performance of several protein structure prediction (PSP) algorithms, including AlphaFold2 and RoseTTAFold, with and without the use of the MSA-Generator. The algorithms are evaluated using various metrics such as pLDDT, LDDT, TM-Score, and GDT-TS. The average depth of the MSAs used in the experiment is also reported.

This table presents the results of evaluating the performance of several protein structure prediction (PSP) algorithms on real-world challenging MSAs. It shows the average pLDDT, LDDT, TM-Score, and GDT-TS scores for each algorithm, along with the improvements achieved by using MSA-Generator. The table compares different methods, including single-sequence-based methods, MSA-based methods (with and without MSA-Generator), and methods that incorporate additional techniques like Potts generation or iterative unmasking. The average depth of the MSAs used is also shown.

This table presents the results of applying various MSA generation methods and evaluating their performance in improving the accuracy of protein structure prediction using Alphafold2 and RoseTTAFold. It shows the average pLDDT, LDDT, TM-Score, and GDT-TS scores across three independent runs, comparing different methods for creating MSAs (including the proposed MSA-Generator) for challenging protein sequences with few homologues.
Full paper#