TL;DR#
Structure-based drug design (SBDD) is crucial for therapeutic development. Existing target-aware generative models, particularly diffusion models, show promise but struggle to generate molecules with consistently high binding affinity and desirable properties, lacking effective control over generation quality. They primarily learn the overall chemical distribution, neglecting to prioritize high-quality candidates.
ALIDIFF, a new alignment framework, directly addresses this. It leverages a preference optimization approach combined with an improved Exact Energy Preference Optimization (E2PO) method to shift the chemical distribution towards molecules with superior binding energies and structural features, as defined by user-specified reward functions. E2PO avoids overfitting and provides a closed-form solution for the converged distribution. ALIDIFF showcases state-of-the-art performance on the CrossDocked2020 benchmark, producing molecules with significantly improved binding energies while maintaining favorable molecular properties.
Key Takeaways#
Why does it matter?#
This paper is important because it presents ALIDIFF, a novel framework to enhance the quality of generated molecules in structure-based drug design. By aligning pre-trained diffusion models with preferred properties, ALIDIFF addresses the limitations of existing models that lack effective steerability. This opens avenues for improving drug discovery by generating molecules with desirable properties, impacting various research fields focusing on drug development and molecular design.
Visual Insights#

🔼 This figure illustrates the core idea of ALIDIFF, a method to align pretrained target-aware molecule diffusion models with preferred functional properties. It shows that ALIDIFF shifts the probability distribution of generated molecules to favor those with higher binding affinity (as measured by a reward function). A protein’s binding pocket is shown, along with a high-reward molecule (well-fitting ligand) and a low-reward molecule (poorly-fitting ligand). The likelihood of generating high-reward molecules is increased, while that of low-reward molecules is decreased.
read the caption
Figure 1: High-level illustration of ALIDIFF. For a protein target, we can have multiple candidate ligands and rank the preference by certain reward functions, e.g., binding energy. We align the target-aware molecule diffusion model with these preferences by adjusting the conditional likelihoods.
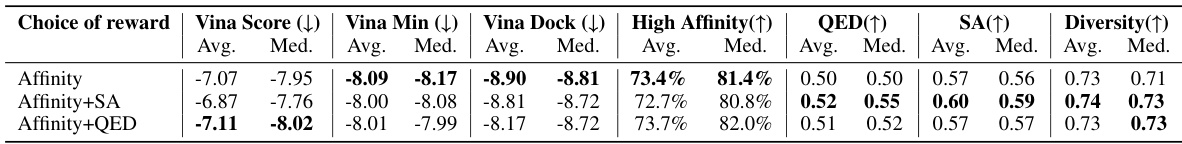
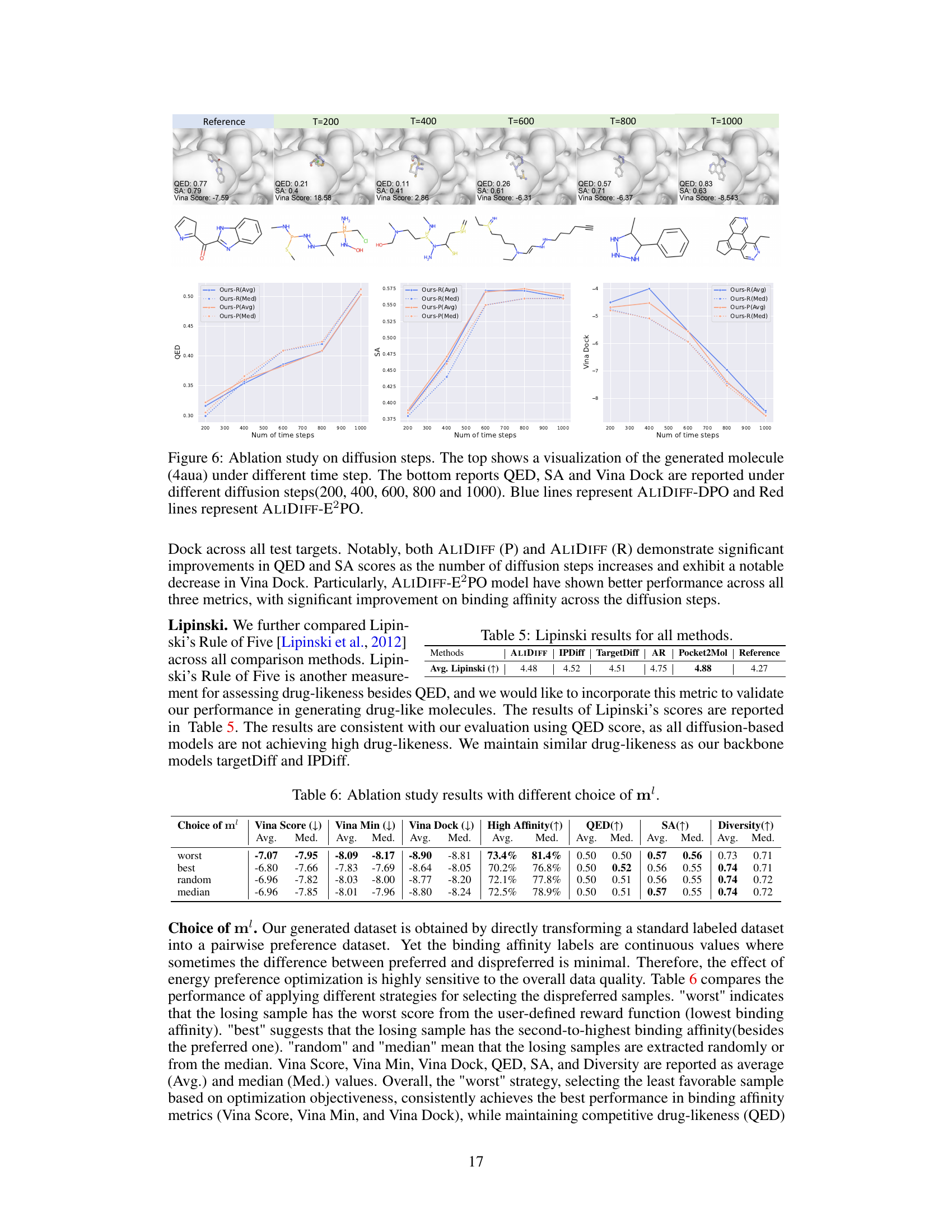
🔼 This table summarizes the performance of ALIDIFF and several baseline models in generating molecules with desirable properties. It compares the binding affinity (Vina Score, Vina Min, Vina Dock, High Affinity) and molecular properties (QED, SA, Diversity) of molecules generated by each method against reference molecules. The metrics are presented as averages and medians, with higher values generally indicating better performance. The arrows (↑/↓) in the header indicate whether higher or lower values are preferred for each metric.
read the caption
Table 1: Summary of binding affinity and molecular properties of reference molecules and molecules generated by ALIDIFF and baselines. (↑) / (↓) denotes whether a larger / smaller number is preferred. Top 2 results are bolded and underlined, respectively.
In-depth insights#
Exact Energy Opt#
The heading ‘Exact Energy Optimization’ suggests a method for aligning target-aware molecule diffusion models with desired properties by precisely controlling the energy landscape. Instead of relying on approximations or heuristics, this approach likely involves a mathematically rigorous method to shift the model’s distribution towards regions with lower binding energies and higher structural rationality. This is a significant improvement over previous methods that primarily focused on learning the chemical distribution of drug candidates without effective control over the quality of generation. The ’exact’ nature emphasizes the accuracy and efficiency of this alignment, potentially avoiding overfitting and providing a closed-form expression for the converged distribution. This would imply a powerful technique that guarantees a precise shift in the model’s output towards the desired molecule characteristics, leading to higher quality, more efficient and targeted molecule generation, thus addressing a key limitation in structure-based drug design. The theoretical foundations and empirical results, therefore, are critical in evaluating its effectiveness and wide applicability.
Diffusion Model#
Diffusion models, in the context of molecular generation, are powerful generative models that learn to gradually add noise to molecular data until it becomes pure noise, and then learn to reverse this process, generating new molecules from noise. This approach allows for the generation of diverse and realistic molecules that might not be easily accessible via traditional methods. A key advantage is their ability to generate molecules in 3D, directly modeling the spatial arrangement of atoms. However, a significant challenge lies in controlling the properties of the generated molecules, such as binding affinity or synthesizability, and current diffusion model approaches may not sufficiently address this. Therefore, aligning pretrained diffusion models with desirable properties is a crucial research direction, necessitating the development of frameworks that effectively steer the generation process toward molecules with specific desired characteristics. Methods incorporating reward functions or preference optimization are promising strategies in this pursuit. Exact energy optimization, for instance, offers a potential solution to overcome the overfitting issue inherent in preference optimization methods, enhancing the efficacy and precision of aligning pretrained models with user-defined properties.
Target Alignment#
Target alignment in machine learning models, particularly generative models, focuses on guiding the model’s output to better match a desired target distribution or specification. This is crucial for applications like drug discovery, where generating molecules with specific properties (e.g., high binding affinity, low toxicity) is paramount. Methods for target alignment often involve incorporating reward functions that quantify desirable properties, which are then used to shape the model’s learning process, shifting the probability mass towards the preferred region of the output space. Challenges arise from the complex interplay between the model’s inherent biases and the reward function’s design, and also the risk of overfitting if the alignment process is not carefully controlled. Effective target alignment necessitates a deep understanding of both the generative model and the target properties, often requiring iterative refinement and careful consideration of the trade-offs involved. Successful alignment leads to more effective and efficient generation of desired outputs, improving overall performance and applicability of machine learning in various fields.
Molecular Properties#
Analysis of molecular properties in drug discovery is crucial for assessing the druggability and feasibility of synthesized molecules. Desired properties often involve a balance between high binding affinity (critical for target interaction) and favorable physicochemical characteristics such as synthetic accessibility, drug-likeness, and stability. High binding affinity is usually prioritized as a primary metric in structure-based drug design, but the impact of molecular properties should not be overlooked. Poor molecular properties can significantly hinder the development process, leading to challenges in synthesis, solubility, absorption, and metabolism. Therefore, a multi-objective optimization approach is often preferred, carefully balancing binding affinity with other essential molecular properties to increase the likelihood of identifying successful drug candidates.
Future Directions#
Future research directions stemming from this work could involve several key areas. First, expanding the range of reward functions beyond binding affinity is crucial. Incorporating properties like synthesizability, toxicity, and metabolic stability would significantly enhance the practical value of generated molecules. This could involve integrating various cheminformatics tools and developing new scoring functions to capture these complex attributes more effectively. Second, addressing the limitations of the current offline learning paradigm is important. The high computational cost of evaluating binding energy currently restricts the approach to offline settings. Exploring online learning strategies, potentially incorporating techniques like reinforcement learning, could enable more efficient and adaptive model refinement. Third, improving the efficiency and scalability of the algorithms would make the method applicable to larger datasets and more complex protein targets. This includes optimizing the preference optimization process and exploring parallelization techniques. Finally, a deeper investigation into the interplay between molecular properties and binding affinity is needed. The current study reveals a trade-off between these; future work should focus on understanding and potentially overcoming this limitation, perhaps through advanced generative modeling techniques or improved reward function design. These are critical steps to transform this promising approach into a powerful tool for practical drug discovery.
More visual insights#
More on figures
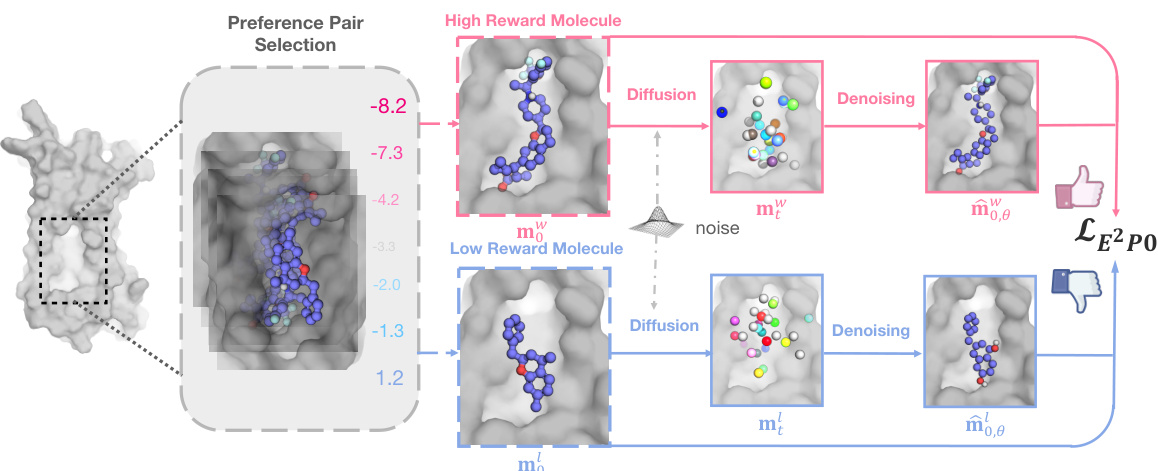
🔼 This figure illustrates the ALIDIFF workflow. For each protein target, two candidate ligand molecules are selected, one with high reward (desirable properties like strong binding energy) and one with low reward. These pairs are used to update the molecule diffusion model via preference optimization, shifting the model’s generation towards higher-quality molecules.
read the caption
Figure 2: Overview of ALIDIFF. This workflow can be summarized as 1) For each protein target (pocket) p in the training set, we retrieve two candidate ligands m; 2) Label the two ligands as winning sample m™ and losing sample m' by desirable properties, e.g., binding energies; 3) Calculate the preference optimization objective Equation (12) and update the molecule diffusion model pe.

🔼 This figure shows the median Vina energy for molecules generated by three different models: TargetDiff, IPDiff, and ALIDIFF, across 100 test samples. The samples are sorted by their median Vina energy as generated by ALIDIFF. The graph visually compares the binding affinity of molecules produced by each model, highlighting the superior performance of ALIDIFF in achieving lower (more favorable) Vina energies. The figure demonstrates ALIDIFF’s ability to generate molecules with significantly improved binding energies compared to the other two methods.
read the caption
Figure 3: Median Vina energy for different generated molecules (TargetDiff, IPDiff, ALIDIFF) across 100 testing samples, sorted by the median Vina energy of molecules generated from ALIDIFF.

🔼 This figure visualizes the generated molecules for two protein pockets (1131 and 2e24) using three different methods: TargetDiff, IPDiff, and ALIDIFF. For each pocket, it shows the reference molecule (the known ligand that binds well to the protein) and molecules generated by each method. The visualization helps compare the structural similarity between the generated molecules and the reference molecule, indicating how well each model is able to generate realistic and effective ligands. The Vina score, QED (quantitative estimate of drug-likeness), and SA (synthetic accessibility) are also shown for each generated molecule, providing a quantitative comparison of the properties of the generated molecules.
read the caption
Figure 4: Visualizations of reference molecules and generated ligands for protein pockets (1131, 2e24) generated by TargetDiff, IPDiff, and ALIDIFF. Vina score, QED, and SA are reported below.
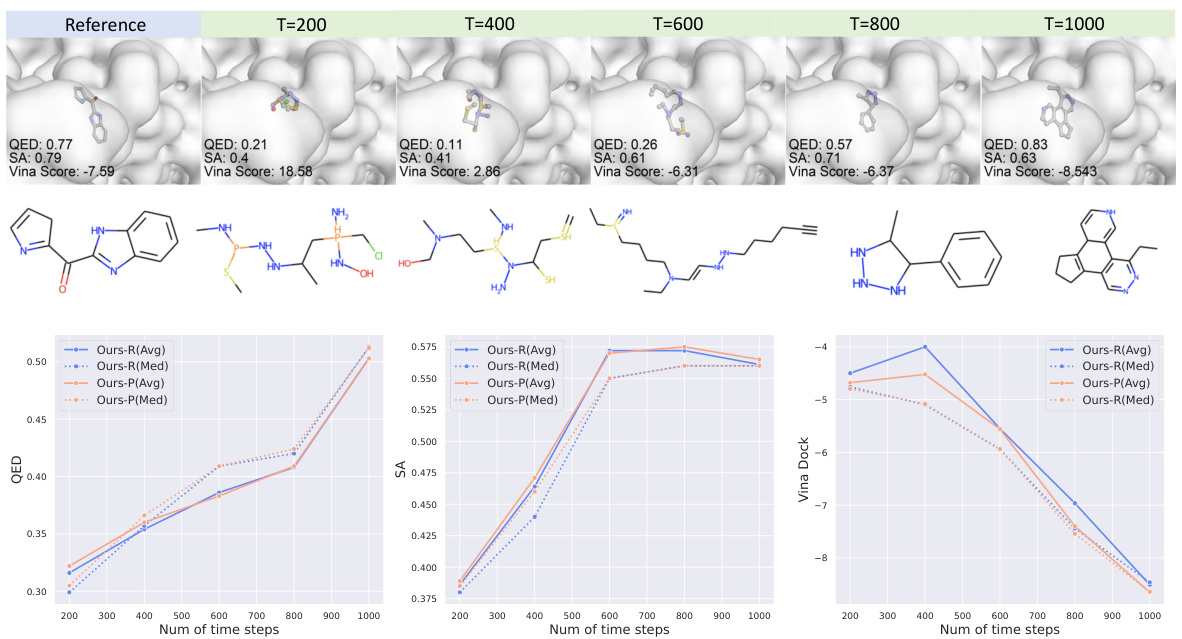
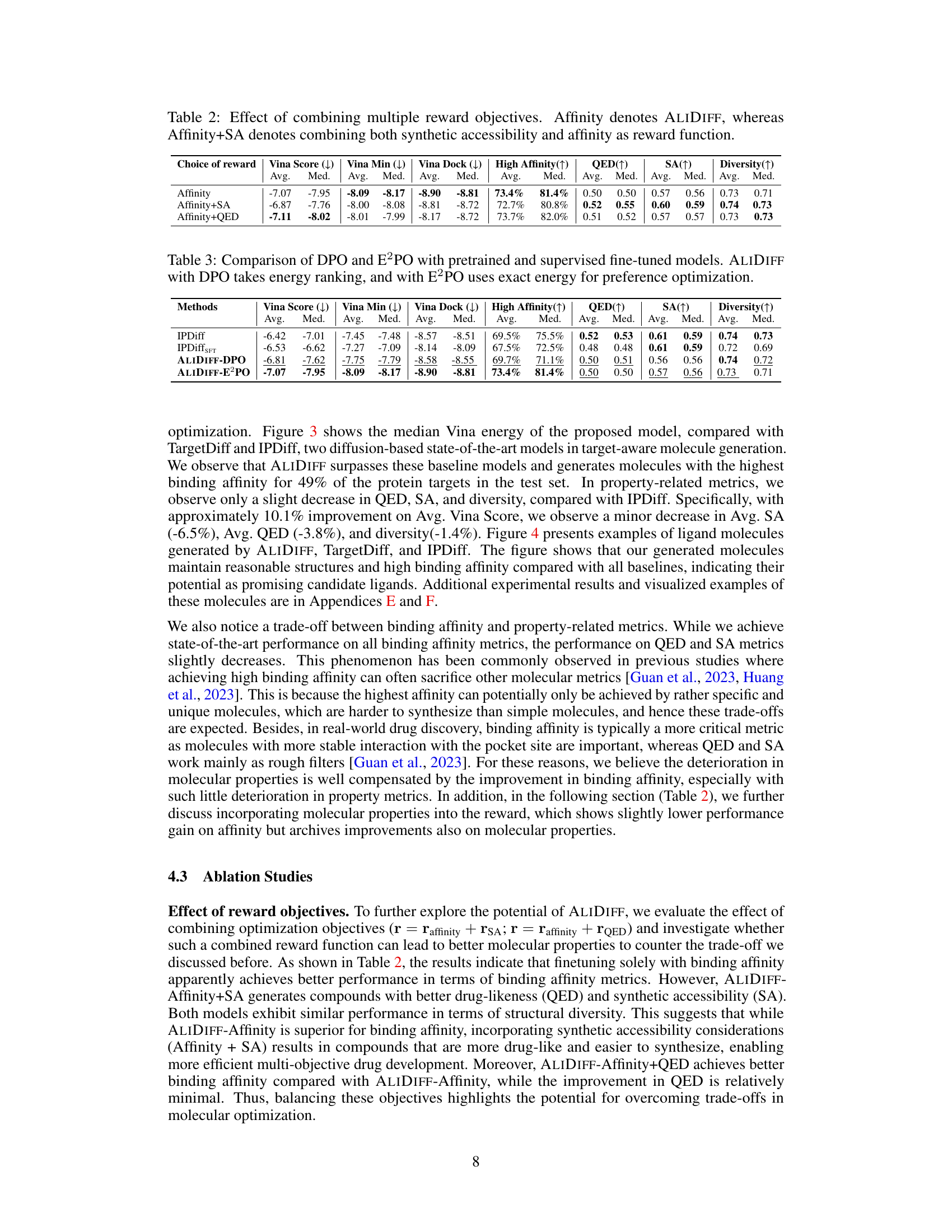
🔼 This figure shows an ablation study on the effect of varying the number of diffusion steps in the ALIDIFF model. The top part displays visualizations of a generated molecule (4aua) at different stages of the diffusion process (200, 400, 600, 800, and 1000 steps). The bottom section presents line graphs illustrating how metrics like QED (drug-likeness), SA (synthetic accessibility), and Vina Dock (binding affinity) change as the number of diffusion steps increases. The results are shown separately for ALIDIFF-DPO (blue lines) and ALIDIFF-E2PO (red lines), highlighting the impact of the exact energy optimization method on these metrics.
read the caption
Figure 6: Ablation study on diffusion steps. The top shows a visualization of the generated molecule (4aua) under different time step. The bottom reports QED, SA and Vina Dock are reported under different diffusion steps(200, 400, 600, 800 and 1000). Blue lines represent ALIDIFF-DPO and Red lines represent ALIDIFF-E2PO.
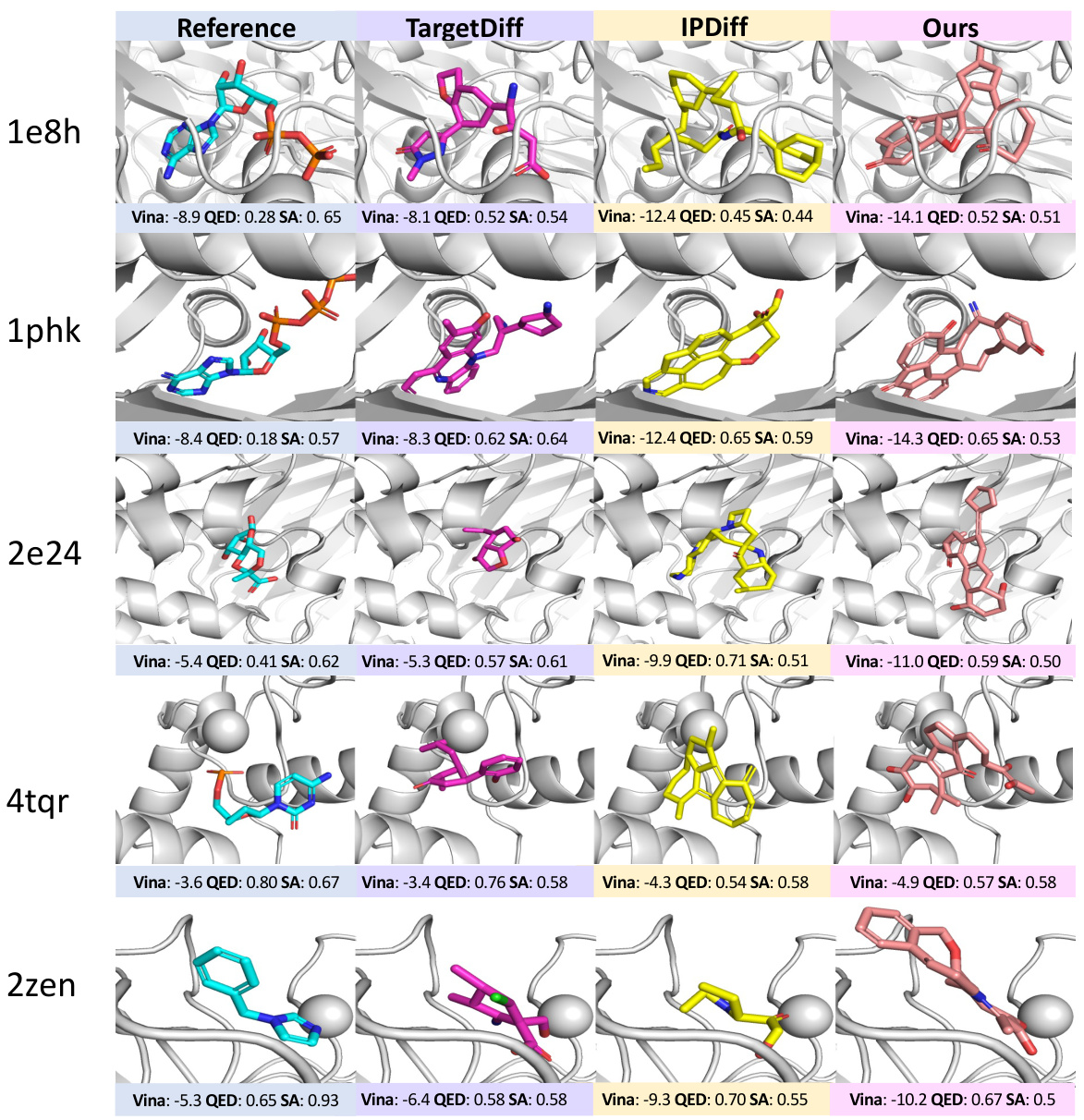
🔼 This figure visualizes the reference molecules and generated ligand molecules for two protein pockets (1131 and 2e24) by three different methods: TargetDiff, IPDiff, and ALIDIFF. For each pocket and method, a representative generated ligand is shown alongside its Vina score (binding affinity), QED (drug-likeness score), and SA (synthetic accessibility score). The figure helps to demonstrate the visual differences and quantitative differences in the quality of the generated molecules produced by the various methods.
read the caption
Figure 4: Visualizations of reference molecules and generated ligands for protein pockets (1131, 2e24) generated by TargetDiff, IPDiff, and ALIDIFF. Vina score, QED, and SA are reported below.
More on tables
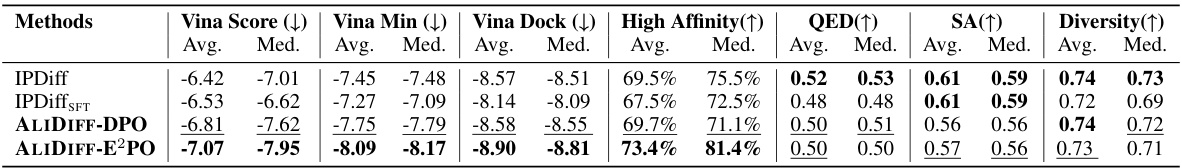
🔼 This table presents a comparison of the binding affinity and several key molecular properties (QED, SA, Diversity, Vina Score, Vina Min, Vina Dock, and High Affinity) of molecules generated by ALIDIFF and several baseline methods. The reference molecules are included as a benchmark for comparison. The upward or downward-pointing arrows indicate whether a higher or lower value is considered more desirable for each property.
read the caption
Table 1: Summary of binding affinity and molecular properties of reference molecules and molecules generated by ALIDIFF and baselines. (↑) / (↓) denotes whether a larger / smaller number is preferred. Top 2 results are bolded and underlined, respectively.
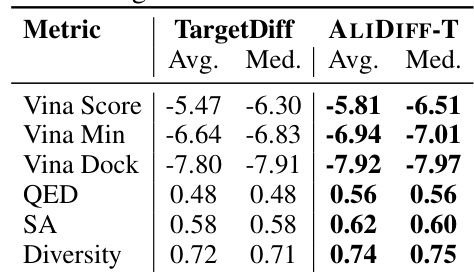
🔼 This table compares the performance of the TargetDiff model with the ALIDIFF-T model (TargetDiff fine-tuned with ALIDIFF) across several metrics. These metrics assess the quality of generated molecules, including binding affinity (Vina Score, Vina Min, Vina Dock), drug-likeness (QED), synthesizability (SA), and diversity. The results demonstrate that ALIDIFF-T improves upon TargetDiff across all metrics, showcasing the effectiveness of the proposed method.
read the caption
Table 4: Finetuning TargetDiff with ALIDIFF. ALIDIFF-T denotes our fine-tuned model with the same reward objective on TargetDiff.

🔼 This table compares the performance of ALIDIFF against several baselines across various metrics related to binding affinity and molecular properties. The metrics include Vina Score (binding affinity), Vina Min (binding affinity after local minimization), Vina Dock (re-docking procedure for optimal binding affinity), High Affinity (percentage of generated molecules with better binding affinity than reference molecules), QED (drug-likeness), SA (synthetic accessibility score), and Diversity. Higher values are preferred for most metrics (denoted by ↑), while lower values are preferred for Vina Score (denoted by ↓). The top two results for each metric are bolded and underlined.
read the caption
Table 1: Summary of binding affinity and molecular properties of reference molecules and molecules generated by ALIDIFF and baselines. (↑) / (↓) denotes whether a larger / smaller number is preferred. Top 2 results are bolded and underlined, respectively.
🔼 This table presents a comparison of the binding affinity and molecular properties of molecules generated by ALIDIFF and several baseline methods. It shows the average and median values for various metrics, including Vina Score, Vina Min, Vina Dock, High Affinity, QED, SA, and Diversity. The upward or downward-pointing arrows indicate whether higher or lower values are preferred for each metric. The top two results for each metric are highlighted.
read the caption
Table 1: Summary of binding affinity and molecular properties of reference molecules and molecules generated by ALIDIFF and baselines. (↑) / (↓) denotes whether a larger / smaller number is preferred. Top 2 results are bolded and underlined, respectively.
Full paper#