TL;DR#
Many machine learning models lack interpretability, hindering their use in science. In the study of gene regulatory networks, understanding the relationships between genes is crucial, but existing neural network models often struggle to reveal this information effectively. Current pruning techniques, while useful for making models computationally efficient, often fail to capture the biologically relevant structure.
This research introduces DASH, a new framework for pruning neural networks. DASH uses available biological knowledge to guide the pruning process, leading to sparser and more easily interpretable models. Experiments on both simulated and real datasets demonstrate that DASH significantly outperforms standard pruning techniques, resulting in models that are both more accurate and more biologically relevant. The method is easily adaptable to other biological modeling applications and potentially other scientific domains.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in machine learning, bioinformatics, and systems biology. It bridges the gap between interpretable models and complex biological systems by introducing a novel pruning technique. This work is relevant to the current trends of interpretable AI and sparse modeling, opening new avenues for research in domain-aware model optimization and biological network inference. The findings have immediate applications for gene regulatory network analysis.
Visual Insights#
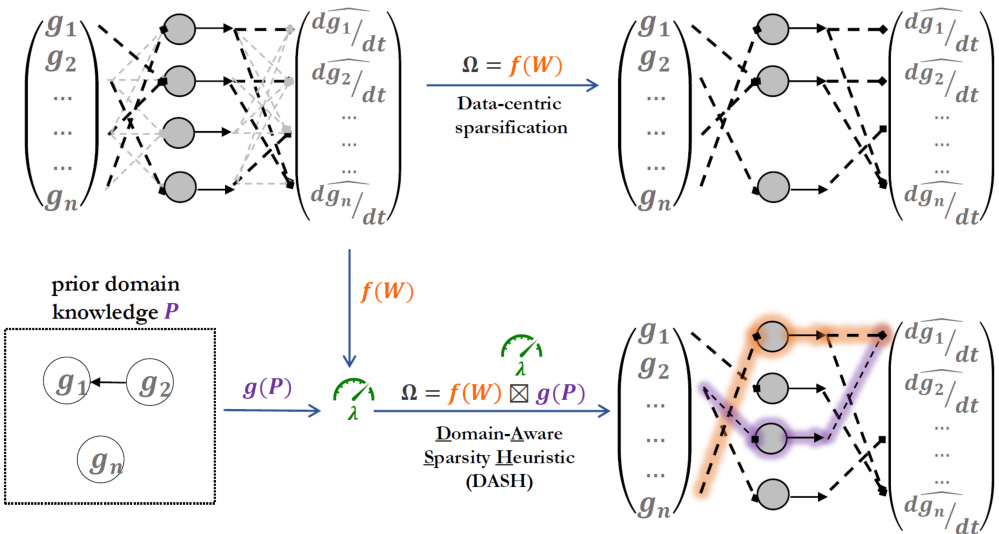
🔼 The figure illustrates the difference between traditional data-centric sparsification and the proposed DASH method. In data-centric sparsification, the pruning score depends only on the learned weights of the neural network. In contrast, DASH incorporates domain-specific knowledge (prior information P) into the pruning score to obtain sparser, more biologically meaningful models. The figure shows a neural ordinary differential equation (NODE) network as an example, where the network learns the gene regulatory dynamics and this information is represented in the weights (W). The input is the expression of a gene, and the output is its derivative.
read the caption
Figure 1: DASH. A NN, here a neural ODE for gene regulatory dynamics, is traditionally sparsified in a data-centric way (top). Pruning is done based on data alone, the pruning score Ω is a function of the learned weights W. Such sparsified models often do not learn plausible relationships in the data domain. We propose DASH (bottom), which additionally incorporates domain knowledge P into the pruning score Ω, yielding sparse networks giving meaningful and useful insights into the domain.

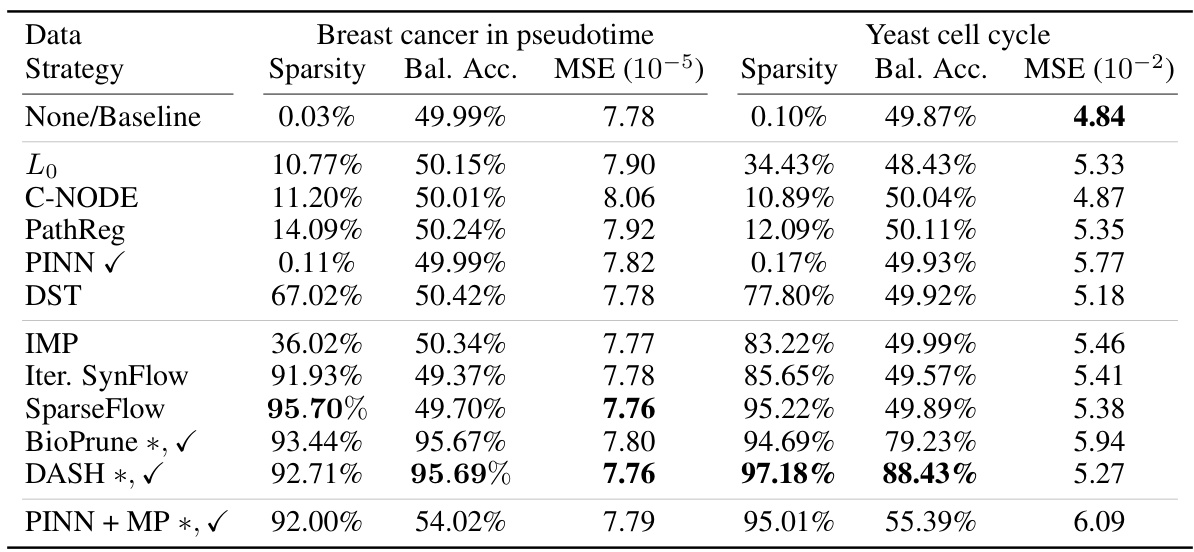
🔼 This table presents a comparison of different gene regulatory network inference methods on synthetic data. It shows the sparsity, balanced accuracy (how well the inferred network matches the true network), mean squared error (prediction accuracy on held-out data), and training time (epochs) for each method. The methods are grouped into those that use penalty-based approaches (implicit pruning), explicit pruning methods, and hybrid approaches. The table highlights the performance of the DASH method in achieving high sparsity and accuracy.
read the caption
Table 1: Synthetic data results. We give model sparsity, balanced accuracy with respect to edges in the ground truth gene regulatory network, mean squared error of predicted gene regulatory dynamics on the test set, and number of epochs (till validation performance plateaus) as proxy of runtime. ✓ is used to indicate methods that leverage prior information. Results are on SIM350 data with 5% noise.
In-depth insights#
Domain-Aware Pruning#
Domain-aware pruning presents a novel approach to neural network pruning by integrating domain-specific knowledge to guide the process. This contrasts with traditional methods that rely solely on data-driven metrics. By incorporating prior information, such as known relationships or structures in the domain, domain-aware pruning aims to improve model interpretability and create models that are not only sparse but also biologically meaningful. The key advantage lies in its ability to produce more robust and accurate models, particularly when dealing with noisy or limited data. This is achieved by constraining the search space for optimal sparse structures, preventing the selection of spurious patterns, often encountered in data-driven methods. The effectiveness of domain-aware pruning is demonstrably shown through its superior performance in recovering ground-truth networks and identifying biologically relevant pathways in gene regulatory network inference, where prior knowledge of biological interactions is effectively leveraged. However, a crucial aspect is the quality and reliability of the prior knowledge, as inaccurate or incomplete information could lead to suboptimal or erroneous results. Future research should focus on addressing this, potentially through techniques that can combine and weigh data and prior knowledge more effectively. This strategy holds immense potential for various scientific domains beyond gene regulatory networks where incorporating domain expertise can improve model accuracy and enhance scientific understanding.
DASH Algorithm#
The proposed DASH algorithm is an iterative pruning method designed for enhancing the interpretability and efficiency of neural networks, particularly in the context of gene regulatory dynamics. DASH uniquely incorporates domain-specific knowledge into the pruning process, unlike traditional data-centric approaches. By integrating prior biological knowledge about gene interactions, DASH guides the pruning procedure to select parameters that align with known relationships, yielding sparser networks that are more biologically plausible. The iterative nature allows for a balance between data-driven and knowledge-driven pruning. Experimental results, using both synthetic and real-world datasets, demonstrate that DASH consistently outperforms other pruning methods in terms of achieving high sparsity while maintaining accuracy and recovering biologically relevant structures. The ability to incorporate domain knowledge is a significant advantage, making DASH applicable to various scientific fields where such knowledge is available.
Gene Regulation#
Gene regulation is a fundamental process governing cellular function, and its intricate mechanisms are critical for normal development and health. Disruptions in gene regulation are implicated in various diseases, including cancer. The research paper explores this complexity by focusing on the inference of gene regulatory networks (GRNs) using neural ordinary differential equations (NODEs). The NODEs offer a powerful approach for modeling dynamic systems, but the resulting models can be challenging to interpret. The paper introduces DASH, a novel framework that combines data-driven techniques with domain knowledge (knowledge about the gene regulatory network itself) to learn sparser and more interpretable NODEs. This domain-aware approach is particularly valuable for biological contexts where biological insights are desired. By incorporating domain knowledge into the model-fitting process, DASH is able to find sparser models that are more biologically plausible and more robust to noise than current state-of-the-art pruning techniques. This approach is shown to be effective across various simulated and real-world datasets, allowing the identification of biologically relevant pathways. DASH therefore demonstrates a significant advance in our ability to model gene regulatory dynamics and gain a deeper understanding of the underlying biological processes.
Sparsity Heuristic#
A sparsity heuristic in the context of neural network pruning for gene regulatory dynamics aims to intelligently identify and remove less important connections within the network. It’s a crucial step that moves beyond simple magnitude-based pruning by incorporating domain knowledge. This knowledge, often in the form of known gene interactions, guides the process, ensuring that the resulting sparse network is not only efficient but also biologically meaningful. A successful sparsity heuristic would balance data-driven optimization with domain-specific constraints, leading to models that are both accurate in their predictions and interpretable in their structure. The challenge lies in finding the right balance—too much reliance on prior knowledge might lead to overfitting, while too little might fail to capture important biological insights. The best approaches would be iterative, allowing the model to adapt to both data and prior knowledge simultaneously, resulting in a sparse model that’s optimized for both predictive accuracy and biological relevance.
Biological Insights#
The concept of “Biological Insights” in the context of a research paper on pruning neural networks for gene regulatory dynamics using data and domain knowledge would focus on how the improved sparsity and interpretability of the pruned models translate into a better understanding of the underlying biological system. DASH (Domain-Aware Sparsity Heuristic), the proposed method, likely aims to uncover meaningful relationships between genes or regulatory elements that were previously obscured by the complexity of the dense neural network. The insights might concern specific regulatory pathways, the roles of key regulatory factors, or the identification of novel interactions. The use of synthetic data with known ground truth helps validate these findings, while real-world data applications demonstrate the capacity of DASH to generate novel biological hypotheses. Comparison with existing methods underscores DASH’s superiority in recovering biologically relevant networks and offers more accurate predictions of gene expression dynamics. The key is that the enhanced interpretability of DASH-pruned models facilitates the extraction of significant biological knowledge, ultimately enabling more precise insights into the complexities of gene regulation.
More visual insights#
More on figures

🔼 This figure compares different pruning strategies on simulated data. The x-axis shows the sparsity of the resulting models (fewer parameters are sparser), and the y-axis displays the balanced accuracy of recovering the true gene regulatory network (higher is better). Methods using prior information are marked with a checkmark. The plot demonstrates that DASH achieves high accuracy while maintaining high sparsity, indicating the model’s effectiveness in recovering true biological relationships.
read the caption
Figure 2: Results on simulated data. We visualize performance of pruning strategies in comparison to original PHOENIX (baseline) in terms of achieved sparsity (x-axis) and balanced accuracy (y-axis) of the recovered gene regulatory network against the ground truth on the SIM350 data with 5% noise. Error bars are omitted when error is smaller than depicted symbol. ✓ indicate methods that leverage prior information. Top left is best: recovering true, inherently sparse biological relationships.
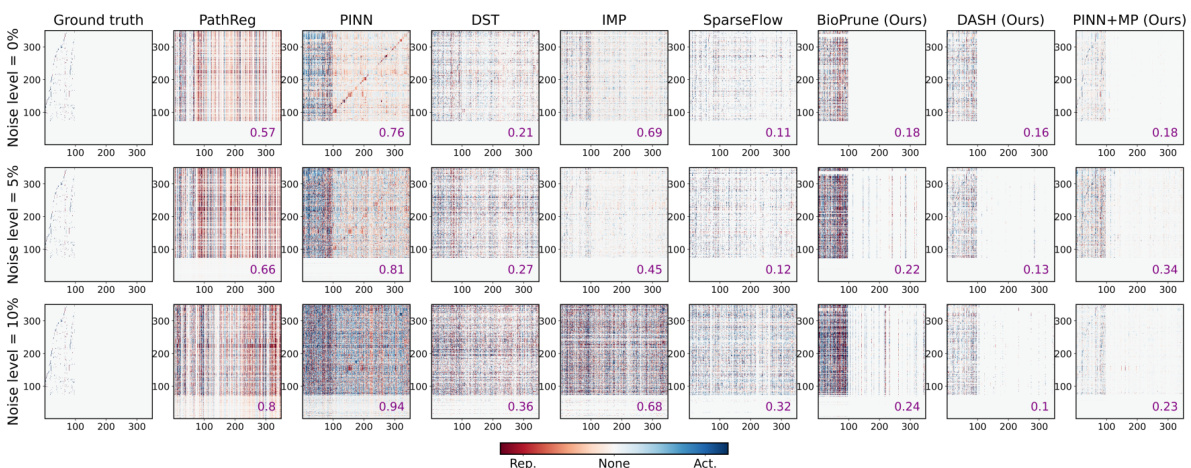
🔼 This figure compares the performance of different gene regulatory network (GRN) inference methods in reconstructing ground truth relationships between genes, at different noise levels. The heatmaps show the estimated effect of one gene on another, with color intensity representing the strength of the effect. The mean squared error (MSE) between the inferred and ground truth networks is shown in purple for each method. The ground truth is shown in the leftmost column, and the methods DASH (Domain-Aware Sparsity Heuristic), BioPrune (a prior-based pruning baseline), and PINN+MP (our suggested method) are shown for comparison in the columns on the right.
read the caption
Figure 3: Reconstruction of ground truth relationships. Estimated effect of gene gj (x-axis) on the dynamics of gene gi (y-axis) in SIM350 for different levels of noise (rows). Ground truth is given on the left, our suggested approach and baselines (DASH, BioPrune, and PINN+MP) on the right with mean squared error between inferred regulatory relationships and ground truth in purple.

🔼 This figure compares various pruning strategies on synthetic gene regulatory network data (SIM350 with 5% noise). The x-axis represents the sparsity (number of remaining parameters) achieved by each pruning method, while the y-axis shows the balanced accuracy of the recovered gene regulatory network compared to the ground truth. Methods that utilize prior biological information are marked with a checkmark. The top-left quadrant represents the ideal scenario: achieving high sparsity with high balanced accuracy, demonstrating successful recovery of the true, sparse biological network structure.
read the caption
Figure 2: Results on simulated data. We visualize performance of pruning strategies in comparison to original PHOENIX (baseline) in terms of achieved sparsity (x-axis) and balanced accuracy (y-axis) of the recovered gene regulatory network against the ground truth on the SIM350 data with 5% noise. Error bars are omitted when error is smaller than depicted symbol. ✓ indicate methods that leverage prior information. Top left is best: recovering true, inherently sparse biological relationships.
🔼 This figure compares the performance of different pruning strategies in reconstructing ground truth relationships between genes in a simulated gene regulatory network. The rows represent different noise levels. The ground truth network is shown on the left, and the results from DASH, BioPrune, PINN+MP, and other methods are displayed on the right. The mean squared error between the inferred and ground truth relationships is shown in purple. It demonstrates the effectiveness of DASH in recovering biologically meaningful relationships even with noise.
read the caption
Figure 3: Reconstruction of ground truth relationships. Estimated effect of gene gj (x-axis) on the dynamics of gene gi (y-axis) in SIM350 for different levels of noise (rows). Ground truth is given on the left, our suggested approach and baselines (DASH, BioPrune, and PINN+MP) on the right with mean squared error between inferred regulatory relationships and ground truth in purple.
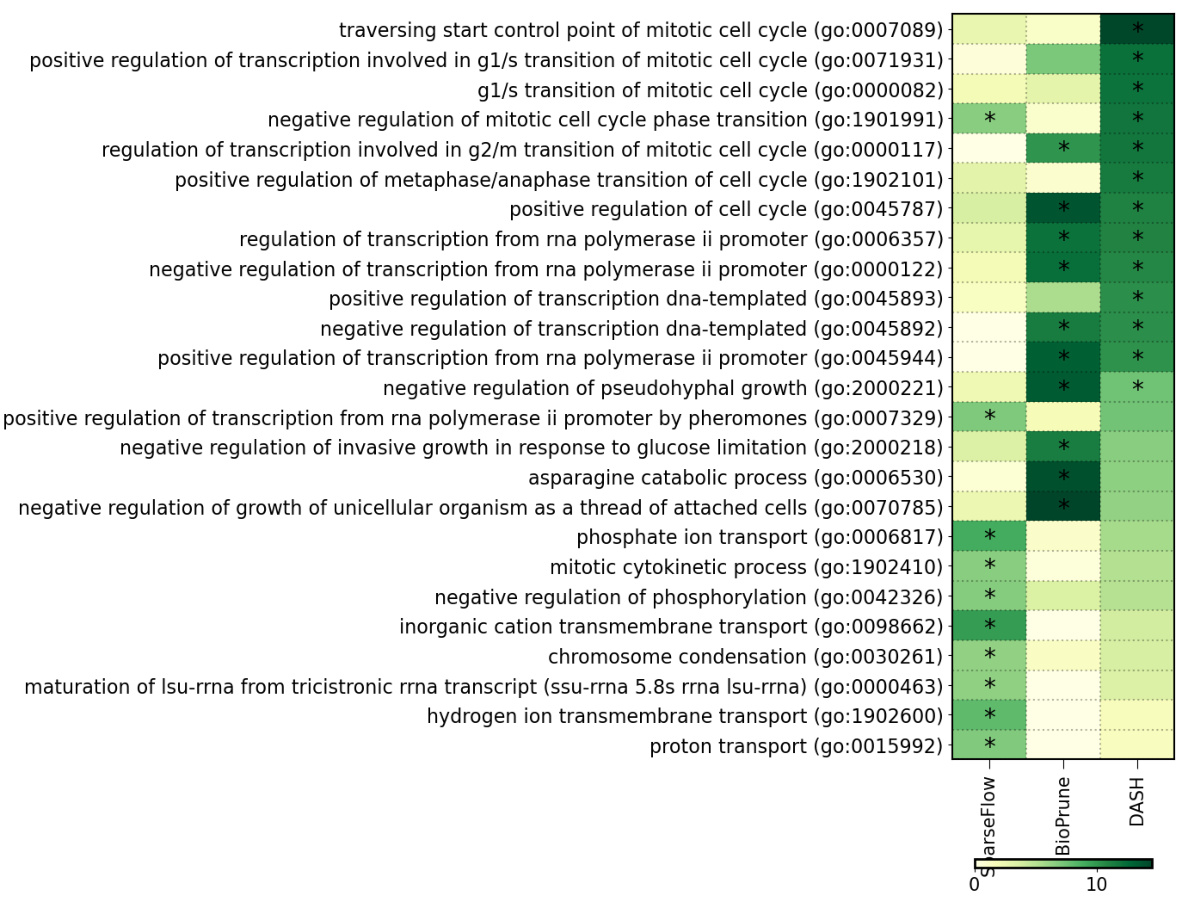
🔼 This figure visualizes the top 20 significant pathways identified by different gene regulatory network inference methods (SparseFlow, BioPrune, and DASH) using yeast data. The x-axis represents the pathway z-score, indicating the statistical significance of each pathway’s enrichment. Asterisks (*) denote pathways that remain significant after correcting for multiple hypothesis testing using the Bonferroni method (a stricter correction for multiple comparisons). The heatmap shows the relative importance of each pathway in each method, offering insights into the biological processes and functions that are most affected by the different network pruning techniques.
read the caption
Figure 6: Yeast pathway analysis. We visualize the top-20 significant pathways for each method, showing the pathway z-score (x-axis) and indicate significant results after FWER correction (Bonferroni, p-value cutoff at .05) with *.

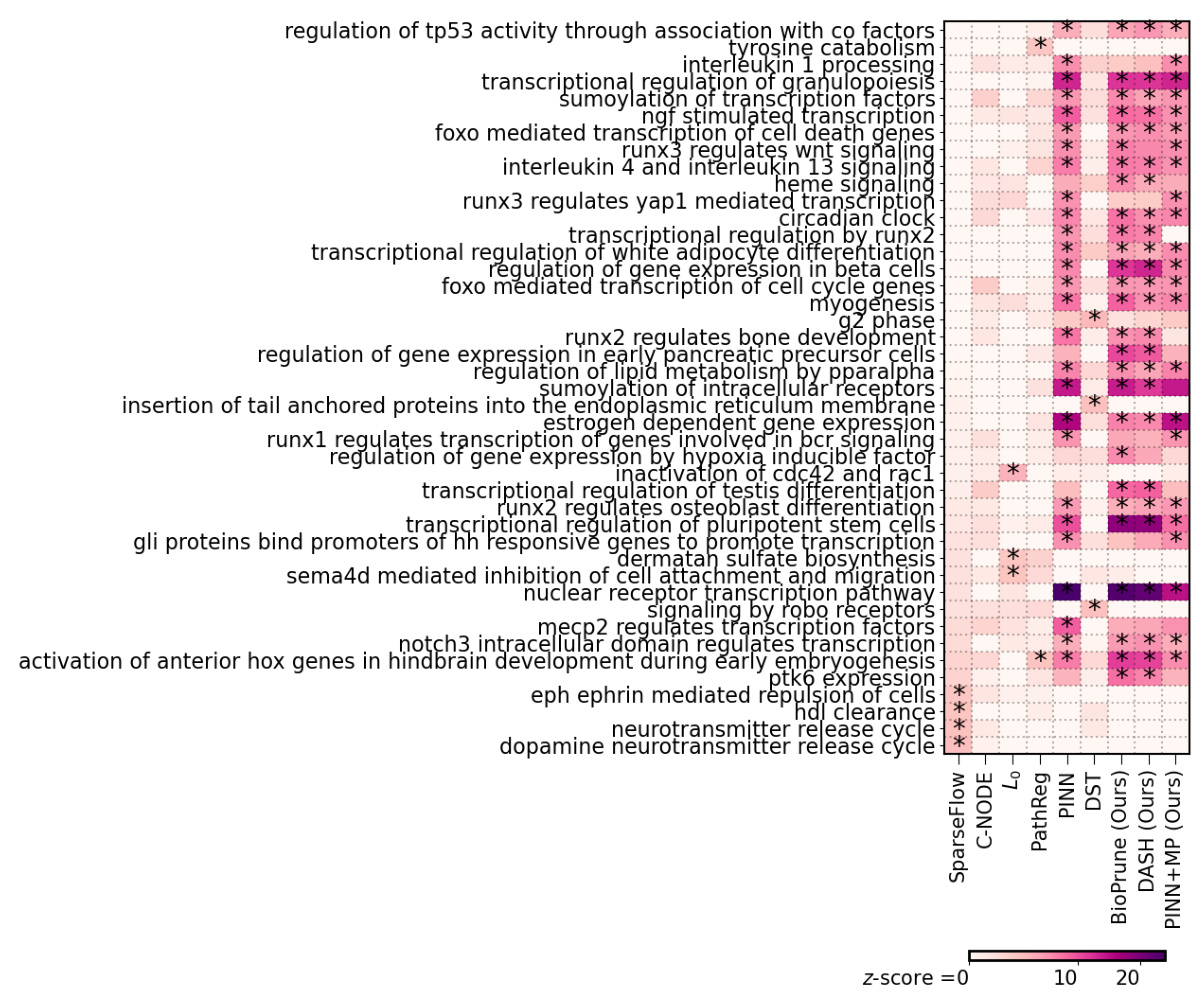
🔼 This figure shows the top 20 significant pathways identified by four different gene regulatory network inference methods (SparseFlow, BioPrune, PathReg, and DASH) applied to breast cancer data. The x-axis represents the z-score, indicating the statistical significance of pathway enrichment. Asterisks (*) denote pathways that remain significant after correcting for multiple hypothesis testing using the Bonferroni method.
read the caption
Figure 5: BRCA pathway analysis. We visualize the top-20 significant pathways for each method, showing the pathway z-score (x-axis) and indicate significant results after FWER correction (Bonferroni, p-value cutoff at .05) with *.
More on tables
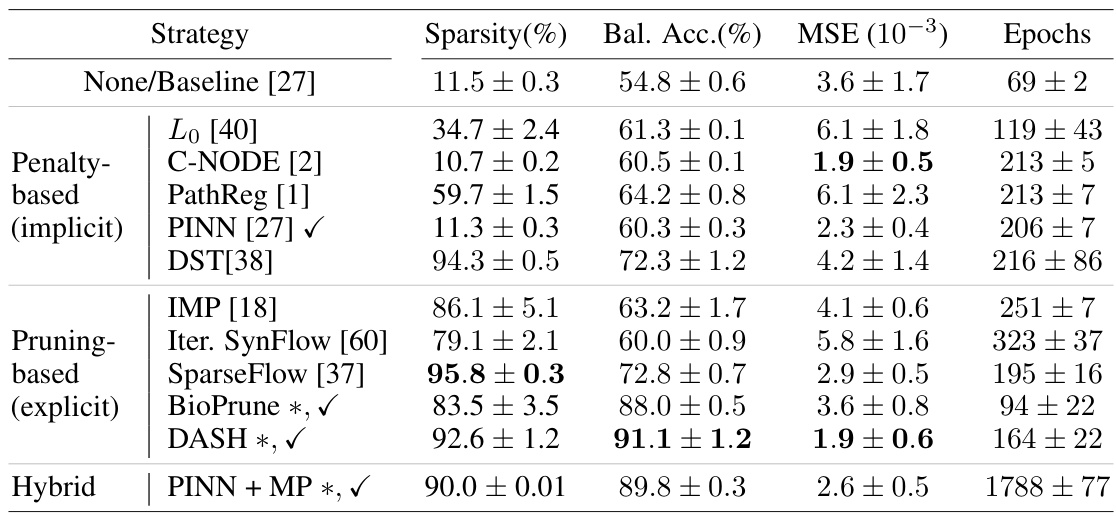
🔼 This table presents the results of applying different gene regulatory network inference methods on breast cancer and yeast cell cycle data. The table shows the sparsity, balanced accuracy (using ChIP-seq data as a gold standard), mean squared error (MSE) of the model predictions, and the number of epochs required until model performance plateaus. The methods are categorized into penalty-based, pruning-based, and hybrid methods, indicating whether or not they leverage prior information. DASH, a domain-aware sparsity heuristic, is compared against these baselines.
read the caption
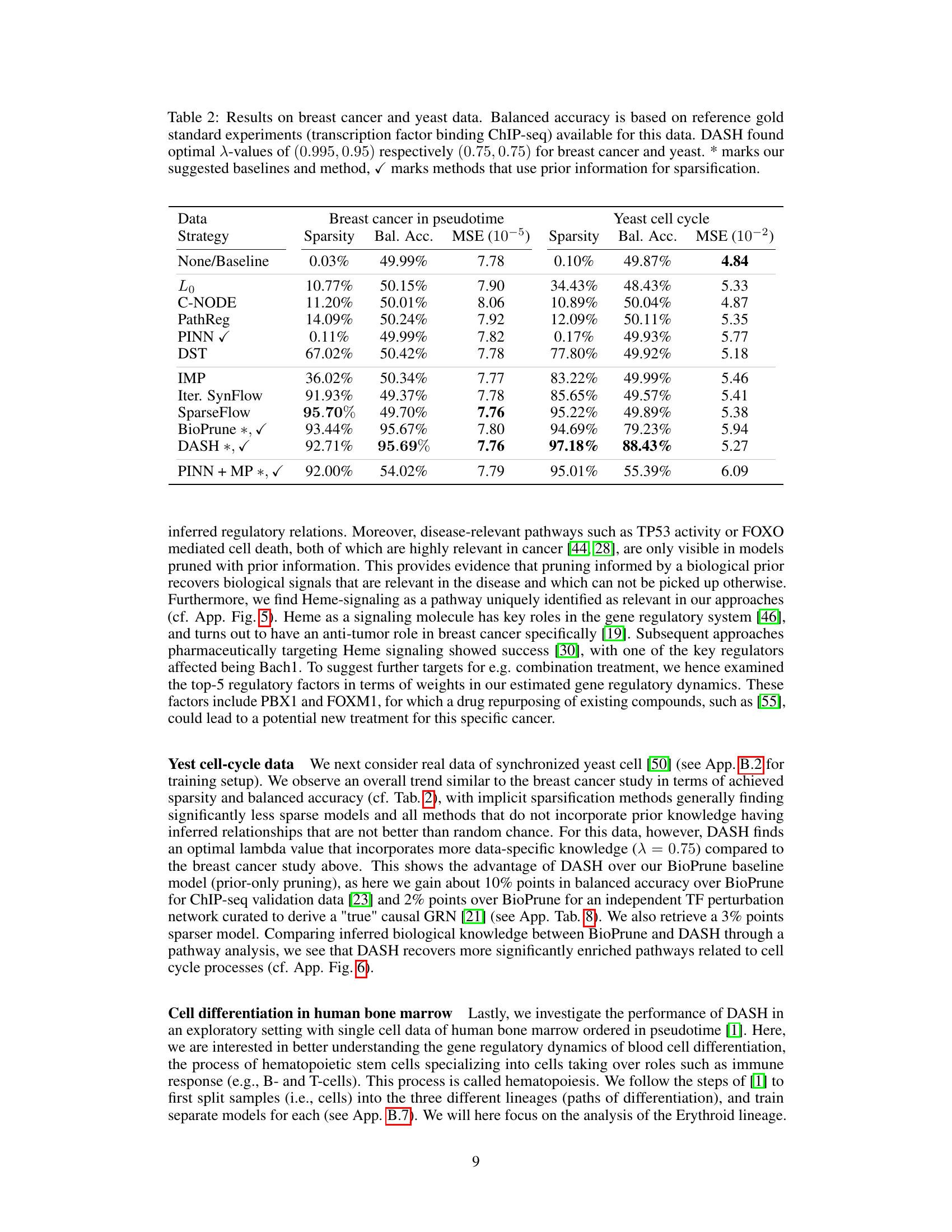
Table 2: Results on breast cancer and yeast data. Balanced accuracy is based on reference gold standard experiments (transcription factor binding ChIP-seq) available for this data. DASH found optimal \textlambda-values of (0.995, 0.95) respectively (0.75, 0.75) for breast cancer and yeast. * marks our suggested baselines and method, \textcheckmark marks methods that use prior information for sparsification.
🔼 This table presents the results of applying various gene regulatory network inference methods on breast cancer and yeast datasets. The table compares the sparsity, balanced accuracy (using ChIP-seq as the gold standard), mean squared error (MSE), and the number of training epochs for each method. Methods that incorporate prior information are marked. DASH achieved high accuracy and sparsity on both datasets.
read the caption
Table 2: Results on breast cancer and yeast data. Balanced accuracy is based on reference gold standard experiments (transcription factor binding ChIP-seq) available for this data. DASH found optimal λ-values of (0.995, 0.95) respectively (0.75, 0.75) for breast cancer and yeast. * marks our suggested baselines and method, ✓ marks methods that use prior information for sparsification.

🔼 This table compares different gene regulatory network inference methods on breast cancer and yeast cell cycle data. It shows model sparsity, balanced accuracy (using ChIP-seq data as a gold standard), mean squared error (MSE) of predictions, and the number of training epochs. The methods are categorized into penalty-based, pruning-based, and hybrid approaches. The table highlights DASH’s performance, especially in achieving high accuracy with a highly sparse model, and shows the impact of using prior information.
read the caption
Table 2: Results on breast cancer and yeast data. Balanced accuracy is based on reference gold standard experiments (transcription factor binding ChIP-seq) available for this data. DASH found optimal λ-values of (0.995, 0.95) respectively (0.75, 0.75) for breast cancer and yeast. * marks our suggested baselines and method, ✓ marks methods that use prior information for sparsification.

🔼 This table presents the results of applying various gene regulatory network inference methods to synthetic data. It compares the performance of different pruning strategies in terms of sparsity, accuracy (balanced accuracy), prediction error (MSE), and training time (epochs). The table highlights the methods that incorporate prior biological information.
read the caption
Table 1: Synthetic data results. We give model sparsity, balanced accuracy with respect to edges in the ground truth gene regulatory network, mean squared error of predicted gene regulatory dynamics on the test set, and number of epochs (till validation performance plateaus) as proxy of runtime. ✓ is used to indicate methods that leverage prior information. Results are on SIM350 data with 5% noise.

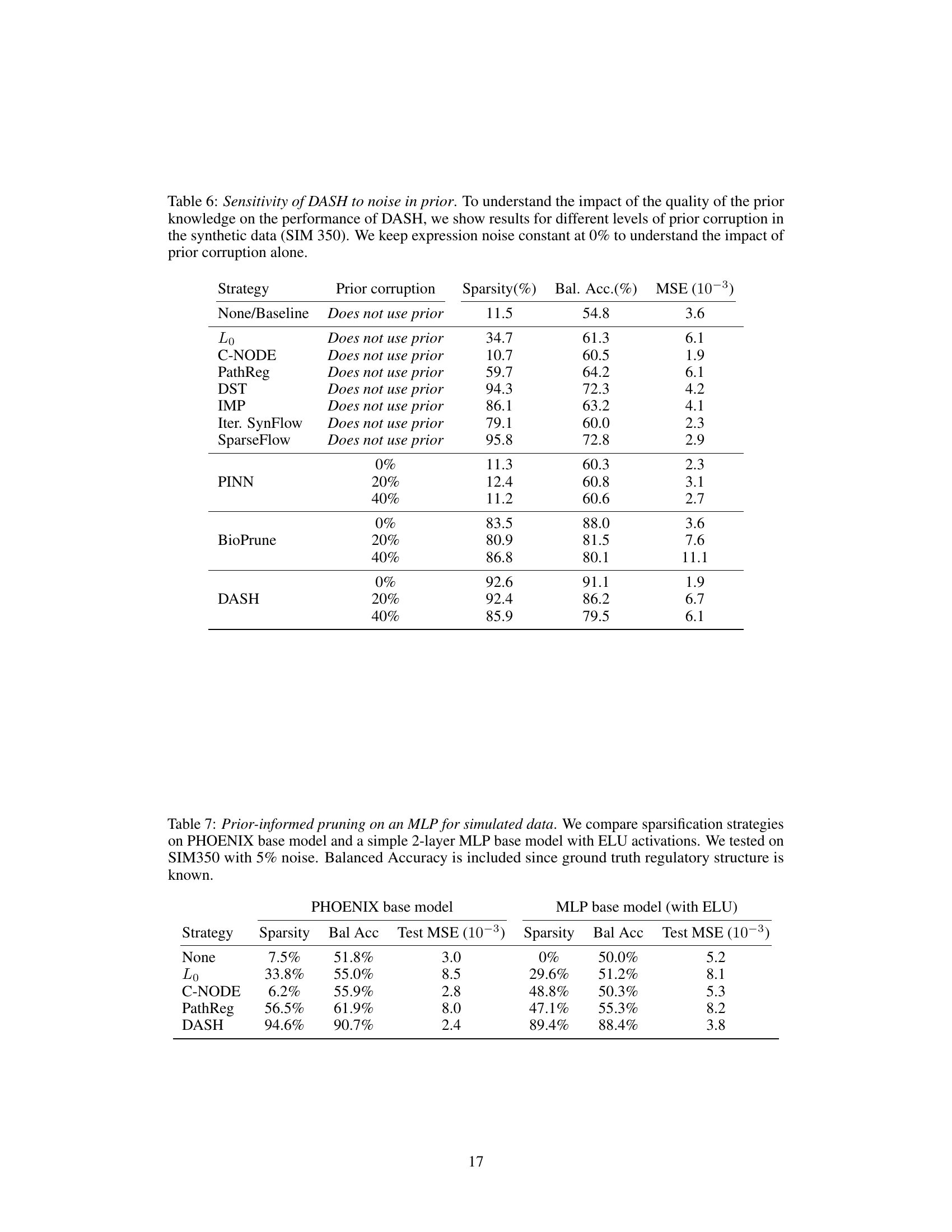
🔼 This table shows the sensitivity analysis of DASH to noise in the prior knowledge. It shows how the performance of DASH (sparsity, balanced accuracy, and MSE) changes with different levels of noise added to the prior knowledge (0%, 20%, and 40%). The expression noise is kept constant at 0% to isolate the effect of prior noise.
read the caption
Table 6: Sensitivity of DASH to noise in prior. To understand the impact of the quality of the prior knowledge on the performance of DASH, we show results for different levels of prior corruption in the synthetic data (SIM 350). We keep expression noise constant at 0% to understand the impact of prior corruption alone.
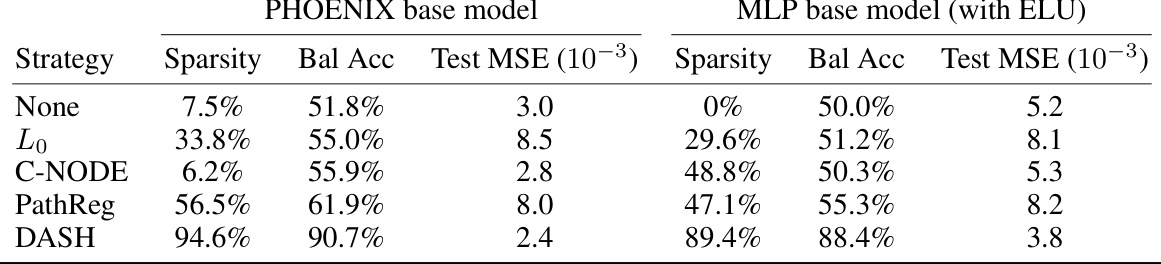
🔼 This table compares the performance of different gene regulatory network pruning methods on real breast cancer and yeast cell cycle datasets. The metrics used are sparsity, balanced accuracy (using ChIP-seq data as a gold standard), mean squared error (MSE) of gene expression prediction, and the number of epochs needed for training. The table also highlights methods that incorporate prior biological knowledge and the optimal hyperparameter values found for DASH.
read the caption
Table 2: Results on breast cancer and yeast data. Balanced accuracy is based on reference gold standard experiments (transcription factor binding ChIP-seq) available for this data. DASH found optimal λ-values of (0.995, 0.95) respectively (0.75, 0.75) for breast cancer and yeast. * marks our suggested baselines and method, ✓ marks methods that use prior information for sparsification.

🔼 This table compares the performance of different gene regulatory network inference methods on breast cancer and yeast cell cycle data. The evaluation metrics include sparsity, balanced accuracy (using ChIP-seq data as a reference), and mean squared error (MSE). The table highlights the superior performance of DASH, particularly in achieving high balanced accuracy and low MSE while maintaining high sparsity. The use of prior information for sparsification is also indicated.
read the caption
Table 2: Results on breast cancer and yeast data. Balanced accuracy is based on reference gold standard experiments (transcription factor binding ChIP-seq) available for this data. DASH found optimal λ-values of (0.995, 0.95) respectively (0.75, 0.75) for breast cancer and yeast. * marks our suggested baselines and method, ✓ marks methods that use prior information for sparsification.

🔼 This table presents the results of applying different gene regulatory network pruning methods to the Erythroid lineage of the Hematopoiesis dataset. The table shows the sparsity achieved by each method, the average out-degree of nodes in the resulting network (as a measure of network complexity), and the mean squared error (MSE) of the gene expression dynamics predictions. Note that a reference gene regulatory network is unavailable, so balanced accuracy cannot be computed. DASH is a method that incorporates prior knowledge into the pruning process and is compared against several other methods. The results show DASH’s ability to achieve high sparsity while maintaining predictive accuracy.
read the caption
Table 9: Results on Hematopoesis data for the Erythroid lineage. We give sparsity of pruned model and test MSE on predicted gene expression dynamics. No reference gene regulatory network is available to compute the accuracy of the recovered network we hence resort to reporting the average out-degree of nodes in the recovered network. DASH found an optimal λ-value of (0.80, 0.80). * marks our suggested baselines and method, ✓ marks methods that use prior information for sparsification.
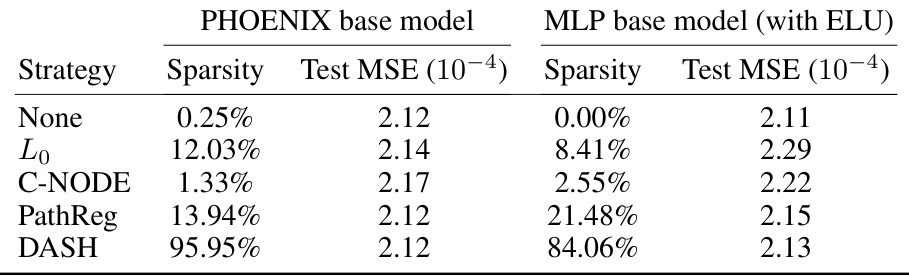
🔼 This table compares the performance of different pruning strategies on two different neural network architectures (PHOENIX and a 2-layer MLP) for the task of modeling gene regulatory dynamics in bone marrow data. The table shows the sparsity achieved and the mean squared error (MSE) for each strategy on the Erythroid lineage. The goal is to determine which pruning strategy produces a sparse network while maintaining good predictive performance. This is an ablation study which shows that the performance of DASH holds up even when changing the base architecture.
read the caption
Table 10: Prior-informed pruning on an MLP for bone marrow data. We compare sparsification strategies on PHOENIX base model and a simple 2-layer MLP base model with ELU activations. We tested on Erythroid lineage of the bone marrow data.
Full paper#