↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Single-cell RNA sequencing (scRNA-seq) is a powerful tool for studying cellular heterogeneity, but the data often suffers from a high proportion of missing values, known as dropouts (false zeros), which significantly hamper downstream analysis. Current imputation methods primarily focus on modeling associating relationships among genes. However, this ignores well-established genetic evidence demonstrating both associating and dissociating relationships among genes.
This paper introduces a novel imputation method called Single-Cell Complete Relationship (scCR) that addresses this issue. scCR constructs a k-NN graph that models both associating and dissociating gene-gene relationships by concatenating the original cell-gene matrix and its negation. The method further standardizes the value distribution of each gene, enhancing the accuracy of propagation. Extensive experiments across six scRNA-seq datasets demonstrate that scCR significantly outperforms state-of-the-art methods in cell clustering and gene expression recovery.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers working with scRNA-seq data because it presents a novel imputation method that significantly improves data quality and downstream analysis results. The proposed scCR method addresses the critical issue of missing values (false zeros) in scRNA-seq data, a common problem hindering accurate analyses. By incorporating both associating and dissociating gene relationships, it enhances accuracy and provides a valuable tool for various single-cell studies. The improved performance in cell clustering and gene expression recovery opens exciting avenues for future research in the field, particularly in improving the understanding of cellular heterogeneity and dynamics.
Visual Insights#

This figure illustrates the two types of relationships that can exist between genes within a single cell. Associating relationships (blue lines) represent genes that tend to co-occur, meaning they are frequently expressed together within the same cell. Dissociating relationships (red lines) represent genes that tend to avoid co-occurrence; when one is expressed, the other is less likely to be expressed. This concept is key to the paper’s proposed method for scRNA-seq data imputation, which incorporates both associating and dissociating gene relationships.
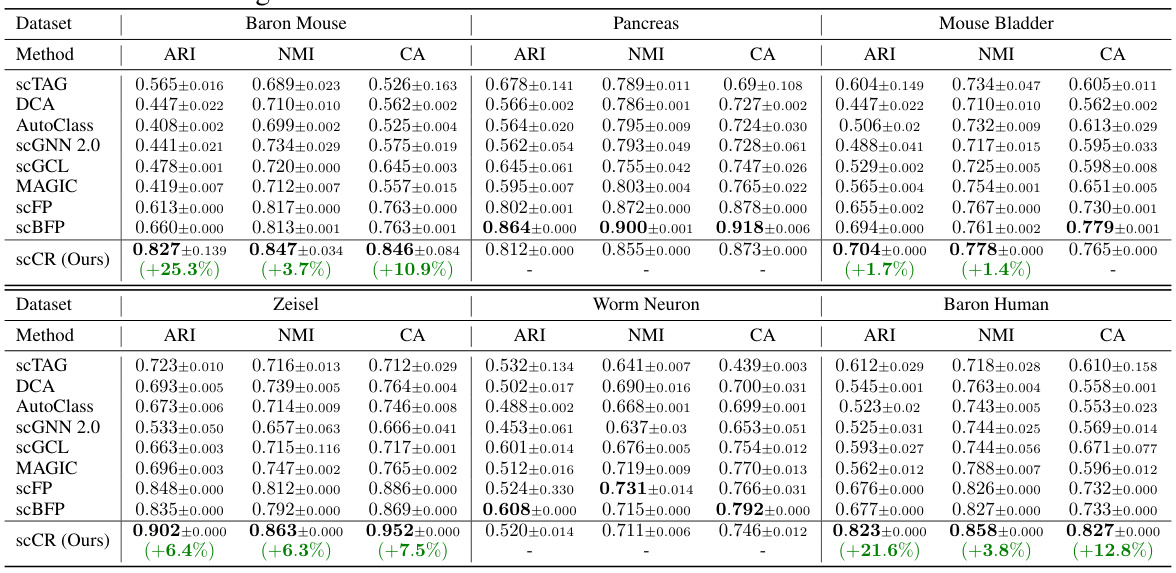
This table presents the performance comparison of the proposed scCR method and eight other state-of-the-art methods for cell clustering on six different scRNA-seq datasets. The performance is measured using three metrics: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). The table highlights improvements of scCR over the best competing method for each dataset.
In-depth insights#
scRNA Imputation#
Single-cell RNA sequencing (scRNA-seq) data is characterized by high levels of dropout, making imputation crucial for downstream analyses. Propagation-based methods, leveraging the relationships between genes, are popular approaches, but often overlook the existence of both associating and dissociating relationships. This paper proposes a novel method, scCR, that addresses this limitation by incorporating both types of relationships. scCR achieves this by concatenating the original cell-gene matrix with its negation, then employing propagation-based imputation on a k-NN graph constructed from this combined and standardized matrix. The standardization step is crucial as it equalizes the varying value distributions among genes, significantly improving imputation accuracy and downstream task performance. The results across various scRNA-seq datasets demonstrate scCR’s superior performance in both cell clustering and gene expression recovery compared to state-of-the-art techniques, highlighting the importance of comprehensive gene-gene relationship modeling in scRNA imputation.
Gene Relation Modeling#
Gene relation modeling in single-cell RNA sequencing (scRNA-seq) data imputation is crucial due to the inherent complexities and noise in the data. Traditional methods often overlook the intricate relationships between genes, focusing primarily on co-expression patterns. A more comprehensive approach considers both associating and dissociating relationships, acknowledging that genes can both positively and negatively influence each other’s expression. This nuanced view requires sophisticated modeling techniques that go beyond simple k-NN graphs to capture the full spectrum of gene interactions. The inclusion of dissociating relationships significantly improves the accuracy of imputation by providing a more complete and biologically realistic picture. Effective imputation is vital for downstream analysis, such as cell clustering and gene expression recovery, making robust gene relation modeling a cornerstone of accurate scRNA-seq data analysis.
scCR Framework#
The scCR framework presents a novel approach to single-cell RNA sequencing (scRNA-seq) data imputation. Its core innovation lies in modeling both associating and dissociating gene-gene relationships, unlike previous methods that primarily focus on associations. This is achieved by concatenating the original cell-gene matrix with its negation, effectively representing both types of relationships. Subsequent standardization ensures that the distribution of each gene’s expression is normalized, preventing biases during propagation. The framework then utilizes a k-NN graph on this standardized, concatenated matrix to propagate information. This propagation step considers both associating and dissociating relationships, refining the imputation and improving downstream analyses. Overall, scCR provides a more comprehensive and biologically grounded imputation method, leading to enhanced performance in cell clustering and gene expression recovery. The framework’s effectiveness is demonstrated through extensive experimentation on several publicly available scRNA-seq datasets.
Dropout Recovery#
The concept of ‘Dropout Recovery’ in single-cell RNA sequencing (scRNA-seq) data analysis focuses on addressing the prevalent issue of missing data, often represented as zero values (dropouts) in the cell-gene expression matrix. These dropouts arise from technical limitations in capturing RNA transcripts and can significantly hinder downstream analyses such as cell clustering and gene expression profiling. Effective dropout recovery methods are crucial for accurate and reliable scRNA-seq data interpretation. Propagation-based imputation methods are widely used to recover dropouts by propagating expression values from similar cells or genes, leveraging the inherent correlation structure in the data. However, these methods often overlook dissociating relationships between genes, which provide additional information for imputation. Advanced methods consider both associating and dissociating relationships for more comprehensive recovery. The effectiveness of various dropout recovery methods is typically evaluated by comparing the imputed data to true expression values using metrics like RMSE (Root Mean Squared Error) and median L1 distance, allowing for a quantitative assessment of the method’s performance.
Future Works#
The paper’s discussion of future work could benefit from a more concrete and detailed exploration of potential research directions. Instead of broadly mentioning extensions to other domains, specific challenges and opportunities should be highlighted. For instance, investigating the impact of different types of noise in scRNA-seq data on the proposed method’s performance would strengthen the paper. Further, it would be beneficial to explicitly address scalability concerns, outlining strategies for handling exceptionally large datasets and the computational requirements. Finally, exploring the biological interpretations of the dissociating relationships, and proposing methods to validate these dissociating relationships experimentally, would be valuable contributions to the field.
More visual insights#
More on figures
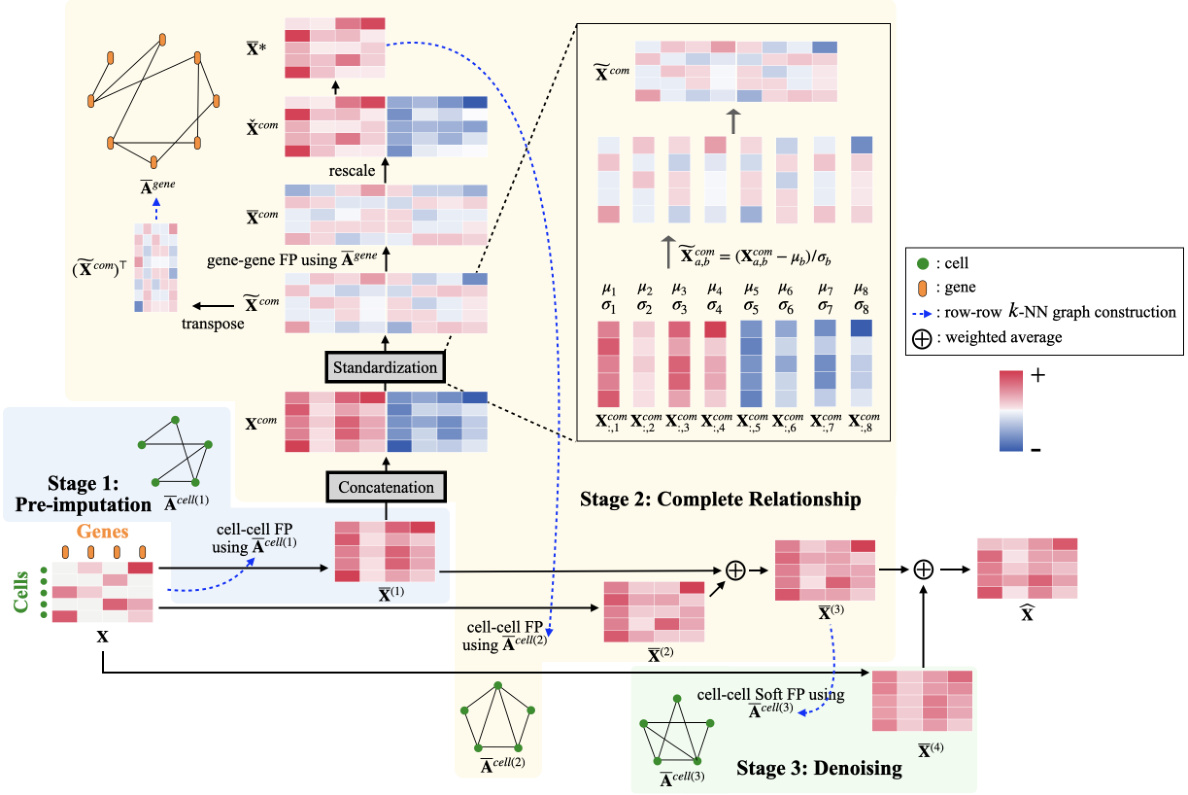
The figure provides a visual overview of the Single-Cell Complete Relationship (scCR) method. It shows the three main stages of the method: pre-imputation, complete relationship, and denoising. Each stage involves specific steps such as k-NN graph construction, feature propagation, and standardization. The figure highlights how the method integrates both associating and dissociating gene-gene relationships to improve imputation accuracy.
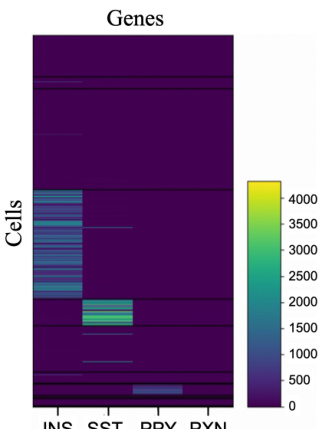
This figure shows a heatmap of a subset of the gene expression matrix from the Baron Human dataset. It visually represents the expression levels of several genes across different cells. The color intensity corresponds to the expression level, with darker shades indicating lower expression and brighter shades indicating higher expression. The figure highlights the variability in gene expression across different genes and cells, illustrating the complexity of single-cell RNA sequencing data and the need for imputation techniques to deal with missing or noisy values.
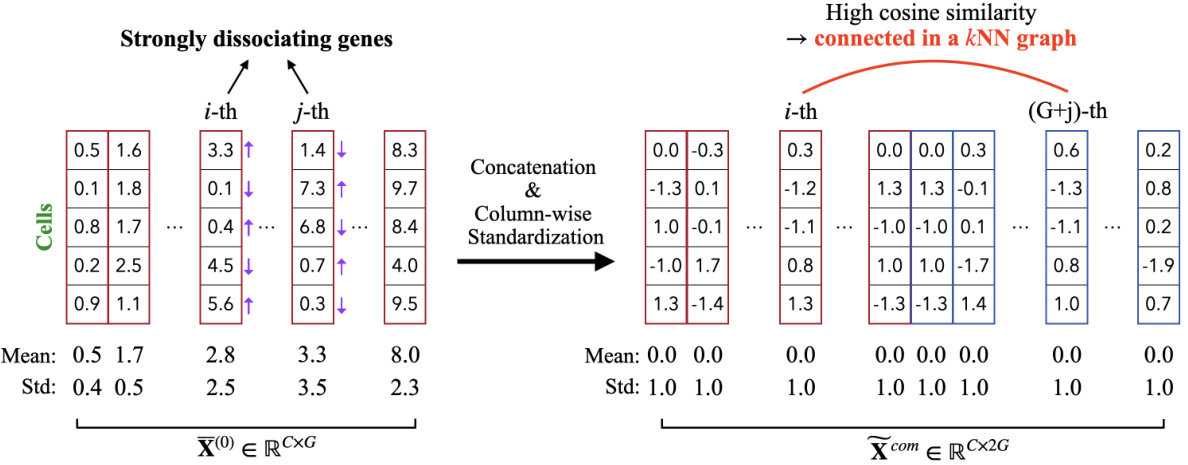
This figure illustrates the two main steps involved in the complete relationship stage of the scCR imputation method. First, concatenation combines the original cell-gene matrix X(1) with its negative counterpart [-X(1)] to create Xcom. This step is crucial for incorporating both associating and dissociating gene relationships. Second, column-wise standardization normalizes the values for each gene in Xcom, ensuring that the data across genes has a standard normal distribution with a mean of 0 and a standard deviation of 1. This process is key for making the subsequent propagation step more effective and preventing the values from various genes dominating the imputation process. The end result is a standardized matrix Xcom, ready for further processing within the scCR framework.
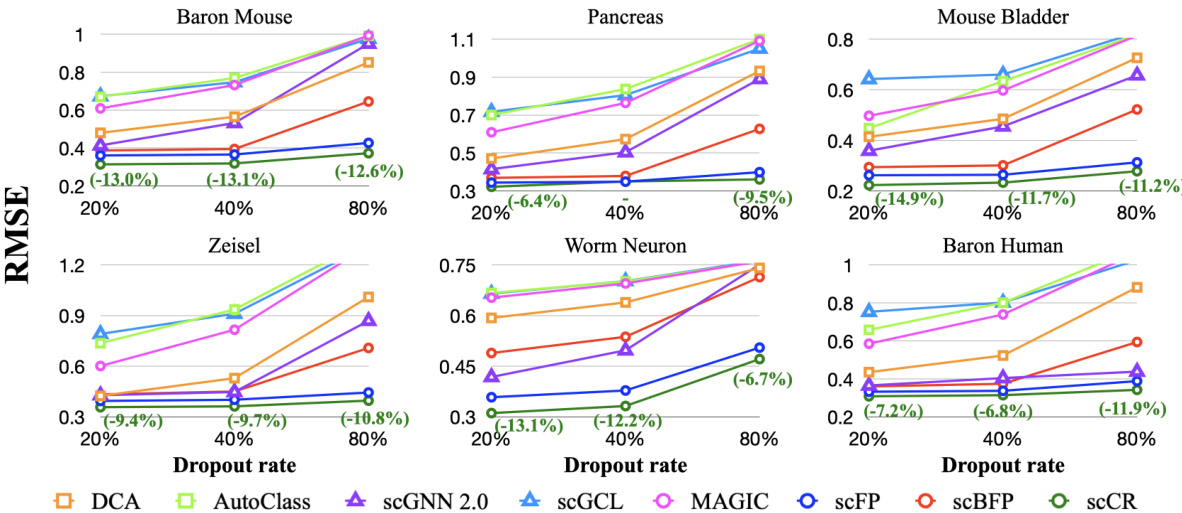
This figure displays the RMSE (Root Mean Square Error) for dropout recovery across six scRNA-seq datasets (Baron Mouse, Pancreas, Mouse Bladder, Zeisel, Worm Neuron, Baron Human) and eight different methods (DCA, AutoClass, scGNN 2.0, scGCL, MAGIC, scFP, scBFP, and scCR). The x-axis represents the dropout rate (20%, 40%, 80%), and the y-axis represents the RMSE. Green highlights indicate that scCR outperforms the best-performing baseline for that specific dataset and dropout rate. The graph shows that scCR consistently achieves lower RMSE values across all datasets and dropout rates, demonstrating its effectiveness in recovering missing data.

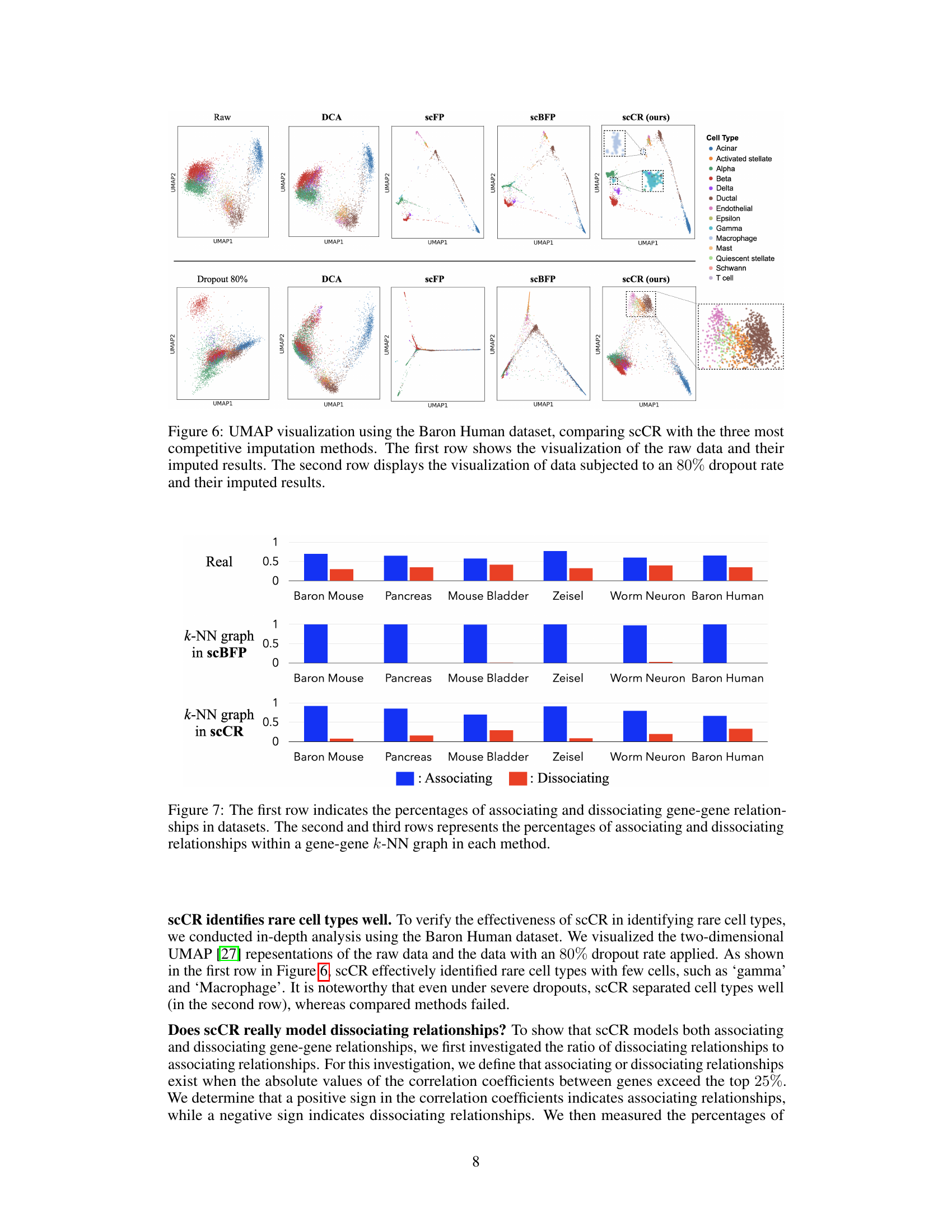
This figure uses UMAP to visualize the results of scCR and three other imputation methods (DCA, scFP, scBFP) on the Baron Human dataset. It compares the visualizations of the raw data with those imputed using each method. A second row shows the same comparison, but with 80% of the data randomly removed (simulating dropouts) before imputation. The visualization allows for a visual comparison of the different methods’ abilities to recover the structure of the data in the presence of noise and missing values.
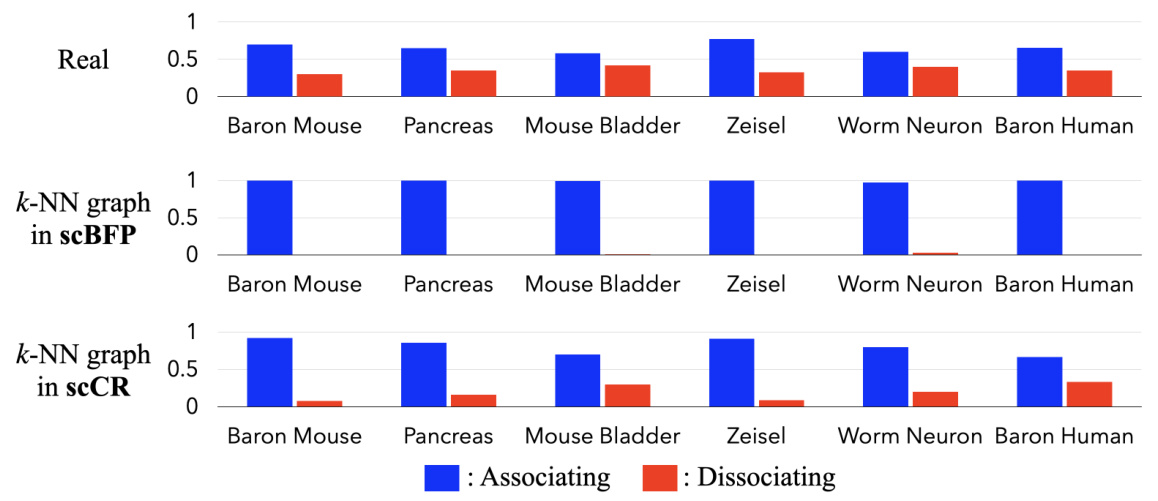
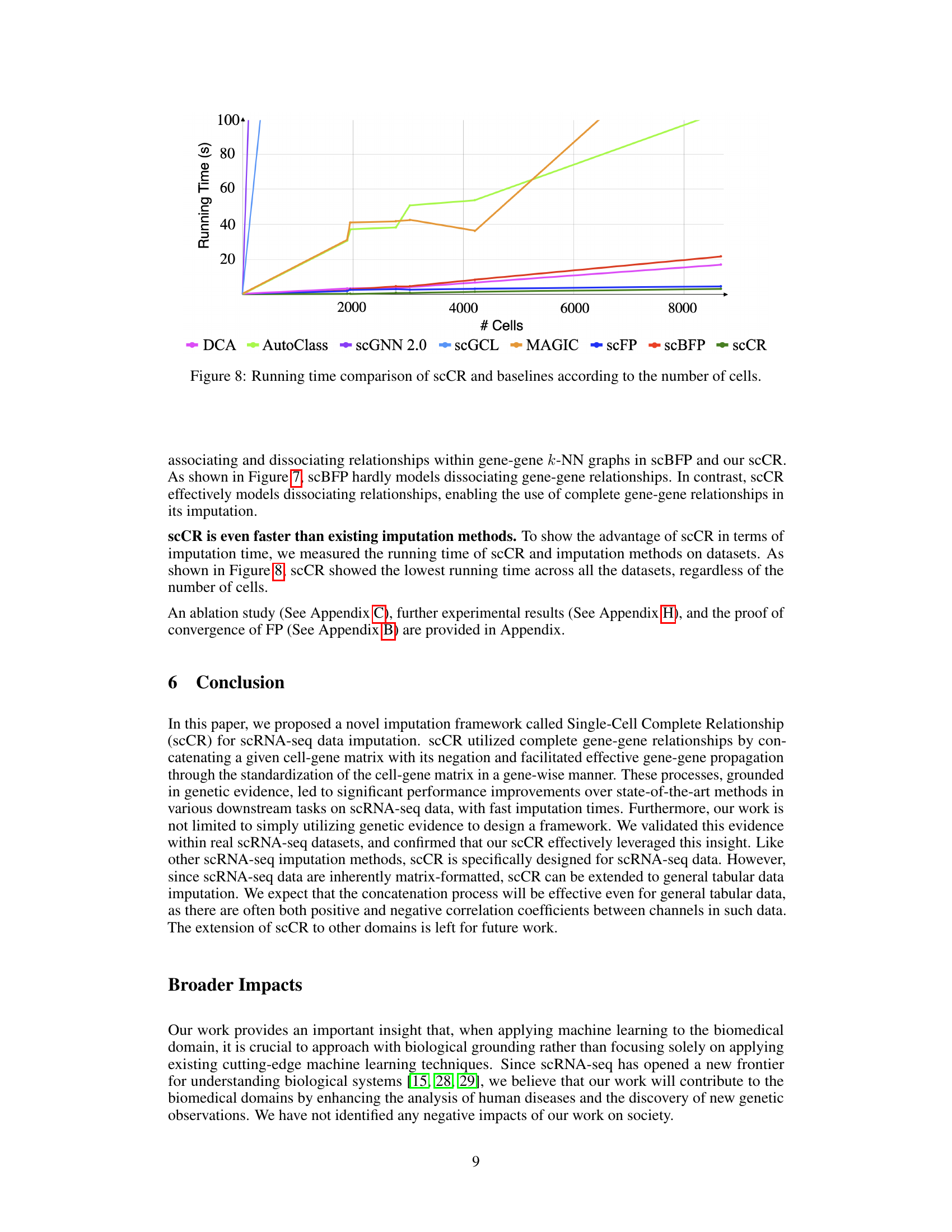
This figure compares the proportion of associating and dissociating gene-gene relationships in six scRNA-seq datasets. It shows the actual ratios in the datasets (first row), and then the ratios captured by scBFP and scCR using their respective k-NN graph constructions (second and third rows). The results highlight scCR’s ability to better capture both associating and dissociating relationships compared to scBFP.
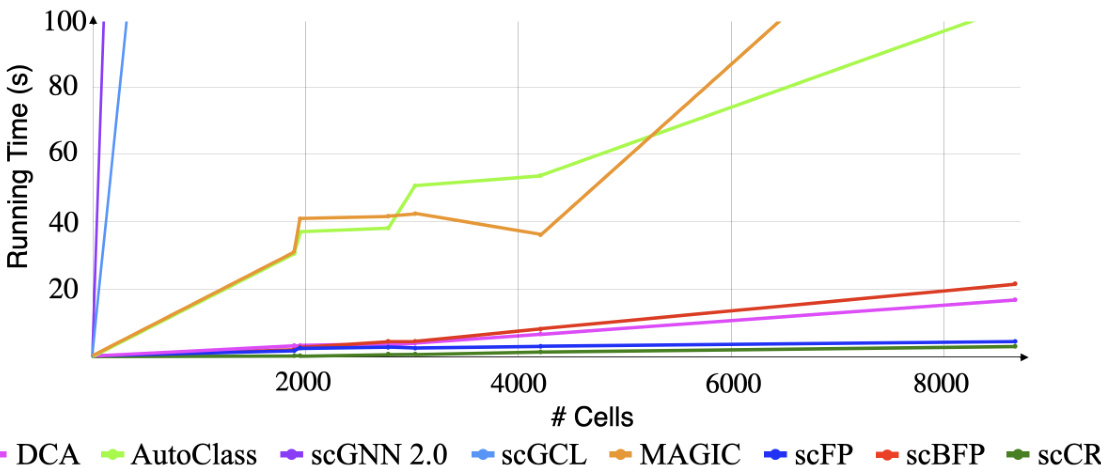
This figure shows the running time of scCR and other state-of-the-art imputation methods (DCA, AutoClass, scGNN 2.0, scGCL, MAGIC, scFP, and scBFP) as a function of the number of cells in the dataset. It demonstrates the computational efficiency of scCR compared to other methods, especially as the number of cells increases. The y-axis represents running time in seconds, while the x-axis represents the number of cells.

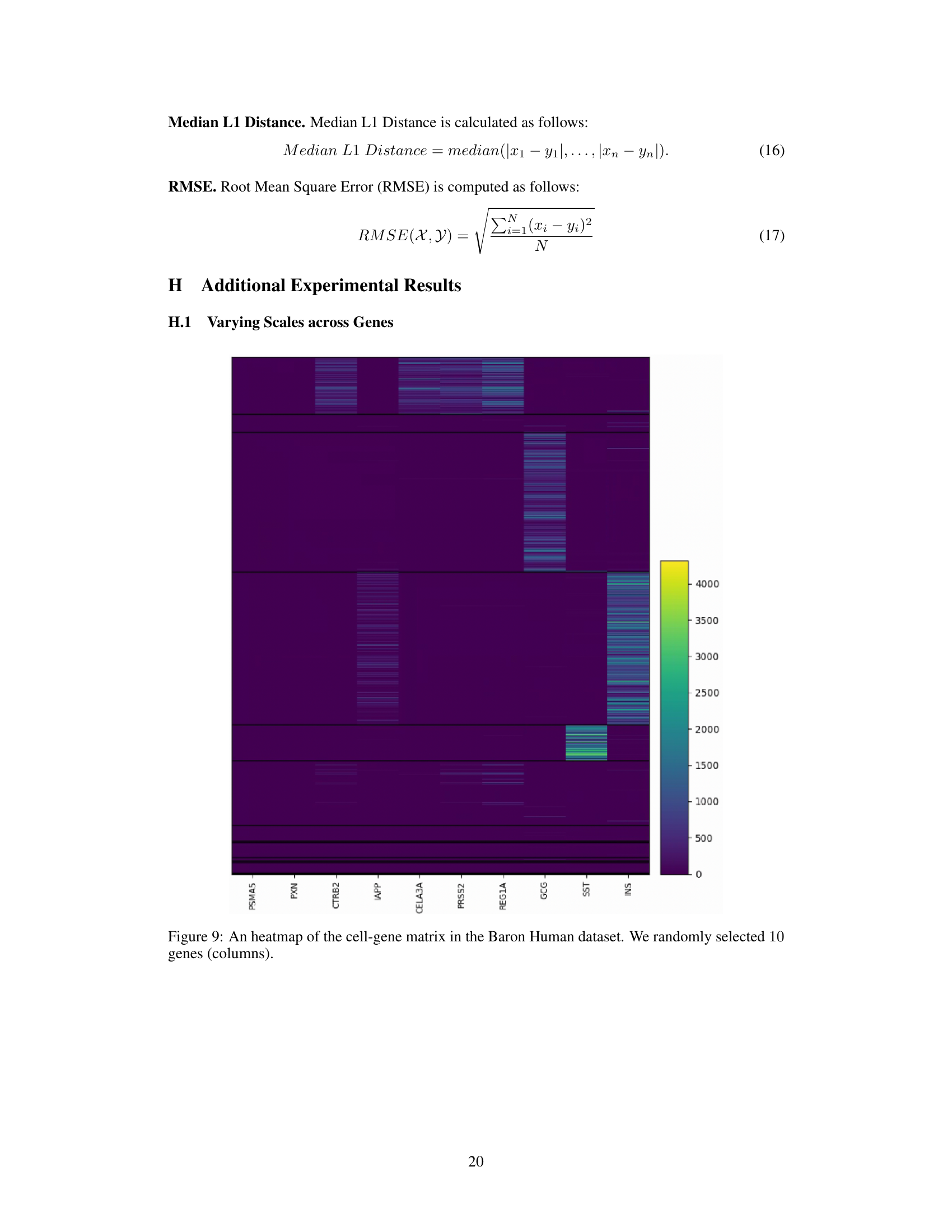
This figure shows a heatmap visualization of a subset of the gene expression matrix from the Baron Human dataset. It highlights the varying scales of gene expression across different genes. The color intensity represents the expression level, demonstrating that the distributions of expression values differ greatly between genes, even within the same dataset. This visual representation emphasizes the heterogeneity of gene expression levels in single-cell RNA sequencing data, which is addressed by the standardization step in the proposed scCR method.

This figure shows the performance comparison of different imputation methods on dropout recovery, measured by the Median L1 Distance metric. The results are shown for six different scRNA-seq datasets (Baron Mouse, Pancreas, Mouse Bladder, Zeisel, Worm Neuron, and Baron Human) and for three different dropout rates (20%, 40%, and 80%). The figure highlights the reduction rates achieved by scCR compared to the best-performing baseline method for each dataset and dropout rate.
More on tables
This table presents the performance of different single-cell RNA-seq imputation methods on cell clustering tasks, evaluated using three metrics: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). The results are shown for multiple datasets. Green highlights indicate improvements over the best performing baseline for each dataset and metric.
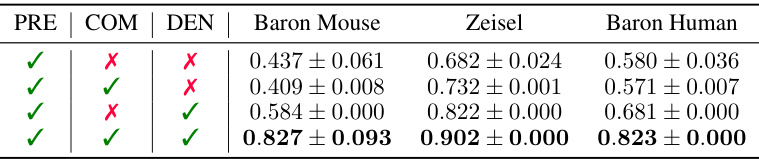
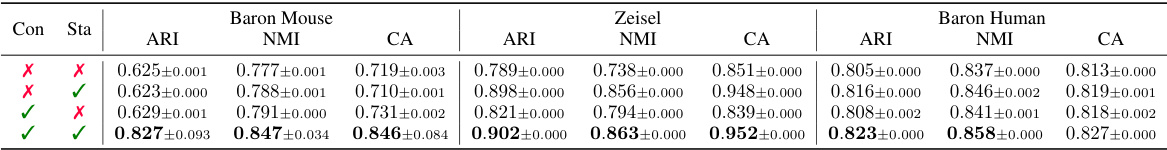
This table presents the results of an ablation study to analyze the effectiveness of each component in scCR. The study was conducted on three datasets: Baron Mouse, Zeisel, and Baron Human. The table shows the performance (ARI, NMI, CA) with different combinations of concatenation (Con) and standardization (Sta). The results demonstrate that both concatenation and standardization significantly contribute to the performance improvement, and their combination leads to the best performance.

This table presents the RMSE (Root Mean Square Error) for dropout recovery under MNAR (Missing Not At Random) conditions. It compares the performance of scFP, scBFP, and the proposed scCR method across six different scRNA-seq datasets (Baron Mouse, Pancreas, Mouse Bladder, Zeisel, Worm Neuron, and Baron Human). Lower RMSE values indicate better performance in recovering the missing values.

This table presents the performance comparison of scCR and eight state-of-the-art methods on six scRNA-seq datasets in terms of cell clustering. Three standard evaluation metrics were used: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). The results show that scCR outperforms other methods in most cases, demonstrating its effectiveness in cell clustering.

This table presents the performance comparison of the proposed method (scCR) and eight state-of-the-art methods on six widely used scRNA-seq datasets. The performance is measured by three standard evaluation metrics: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). The table highlights the improvements of scCR over the most competitive baseline for each dataset.
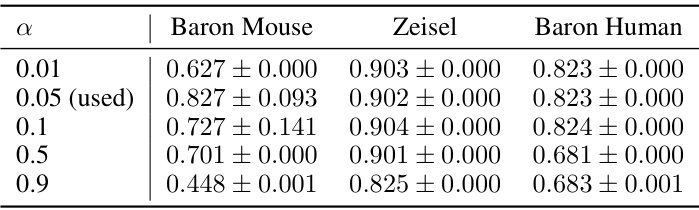
This table presents the performance of scCR and other state-of-the-art methods on six scRNA-seq datasets in terms of cell clustering. The performance is evaluated using three metrics: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). Standard deviation errors are reported, and improvements over the best performing baseline are highlighted.
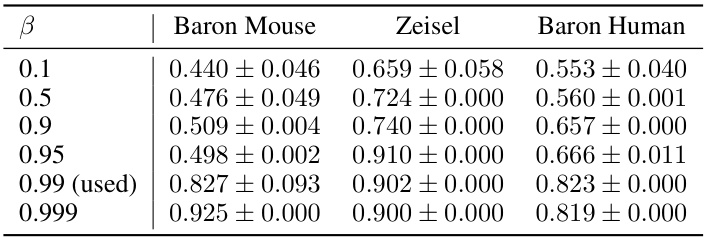
This table presents the performance of scCR and other state-of-the-art methods on six scRNA-seq datasets for cell clustering. The performance is evaluated using three metrics: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). Standard deviations are included to show the variability in the results. Improvements over the best baseline method are highlighted in green.
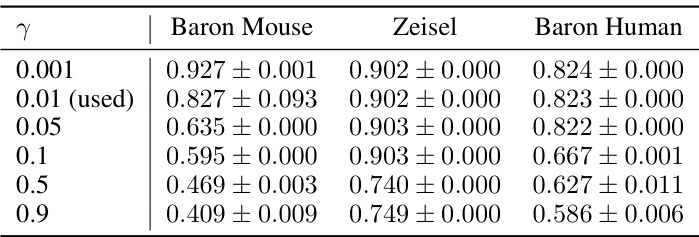
This table presents the performance comparison of the proposed method (scCR) and eight state-of-the-art methods on six scRNA-seq datasets for cell clustering. Three metrics are used: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). The results show that scCR outperforms the baselines in most cases, indicating the effectiveness of the complete gene-gene relationship modeling approach.
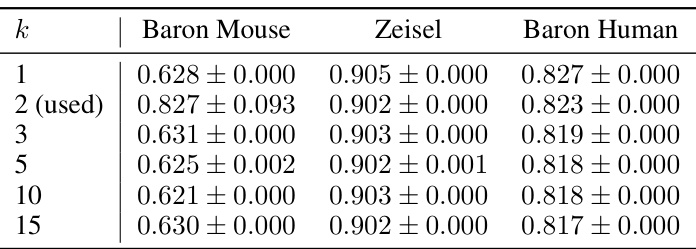
This table presents the performance of scCR and other state-of-the-art methods on six different scRNA-seq datasets in terms of cell clustering. Three metrics are used for evaluation: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). The results show scCR’s improvements over existing methods, highlighting its effectiveness in cell clustering tasks.

This table presents the performance comparison of scCR and other state-of-the-art methods on six different datasets in terms of cell clustering. The performance is measured using three metrics: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). Standard deviation errors are also provided for each result. Values highlighted in green indicate that scCR outperforms the best baseline method for that particular dataset and metric.

This table presents the performance of scCR and eight other state-of-the-art methods on six scRNA-seq datasets for cell clustering. The performance is evaluated using three metrics: Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Clustering Accuracy (CA). Standard deviation errors are included to show the variability in the results. Improvements over the best-performing baseline method are highlighted in green for better readability.
Full paper#