↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Molecular representation learning (MRL) is critical for drug discovery and materials science, but existing contrastive learning (CL) methods often suffer from inaccurate positive and negative pair assignments due to graph augmentations. This leads to suboptimal performance. Many existing works attempt to solve this with heuristics, but this paper addresses the root issue.
This paper introduces a novel probability-based CL framework. It uses Bayesian modeling to learn a weight distribution for each pair, automatically mitigating false pairs. The model is trained via stochastic expectation-maximization. Experiments show significant improvements over existing methods across multiple benchmarks, achieving new state-of-the-art performance on average in multiple datasets.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in molecular representation learning as it presents a novel solution to a critical problem—the prevalence of false positive and negative pairs in existing contrastive learning methods. Its introduction of a probability-based framework and the use of Bayesian inference provides a more robust and accurate method for learning representations, potentially advancing drug discovery and materials science. The detailed experimental results and ablation studies provide valuable insights and benchmarks for future research. This work also opens exciting new avenues for research by combining Bayesian methods with contrastive learning.
Visual Insights#
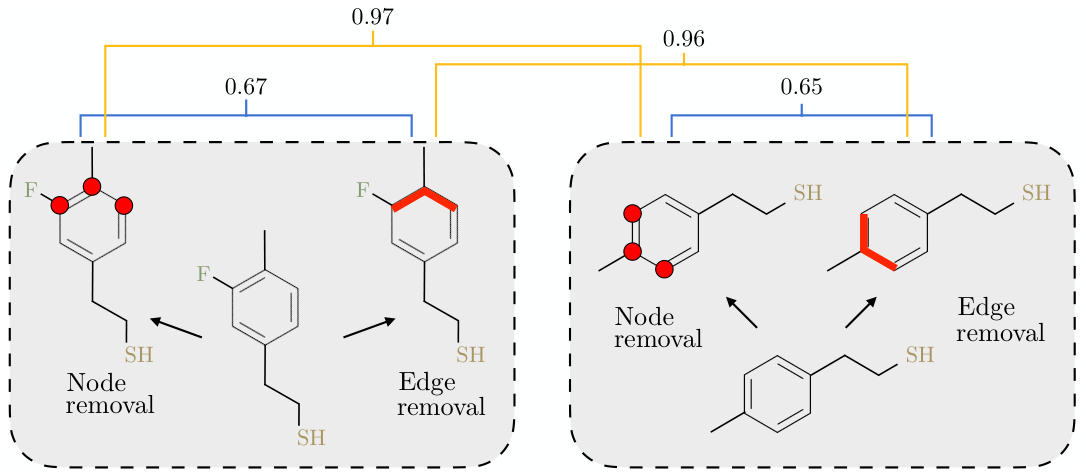
This figure illustrates a common problem in molecular contrastive learning where standard augmentation techniques like node and edge removal can lead to inaccurate labeling of positive and negative pairs. The example shows that molecules with similar chemical structures (high similarity scores) can be incorrectly labeled as negative pairs due to the augmentations, while molecules with lower similarity might be incorrectly labeled as positive pairs. This highlights the need for a more robust method that accounts for such uncertainties in data pair assignments.

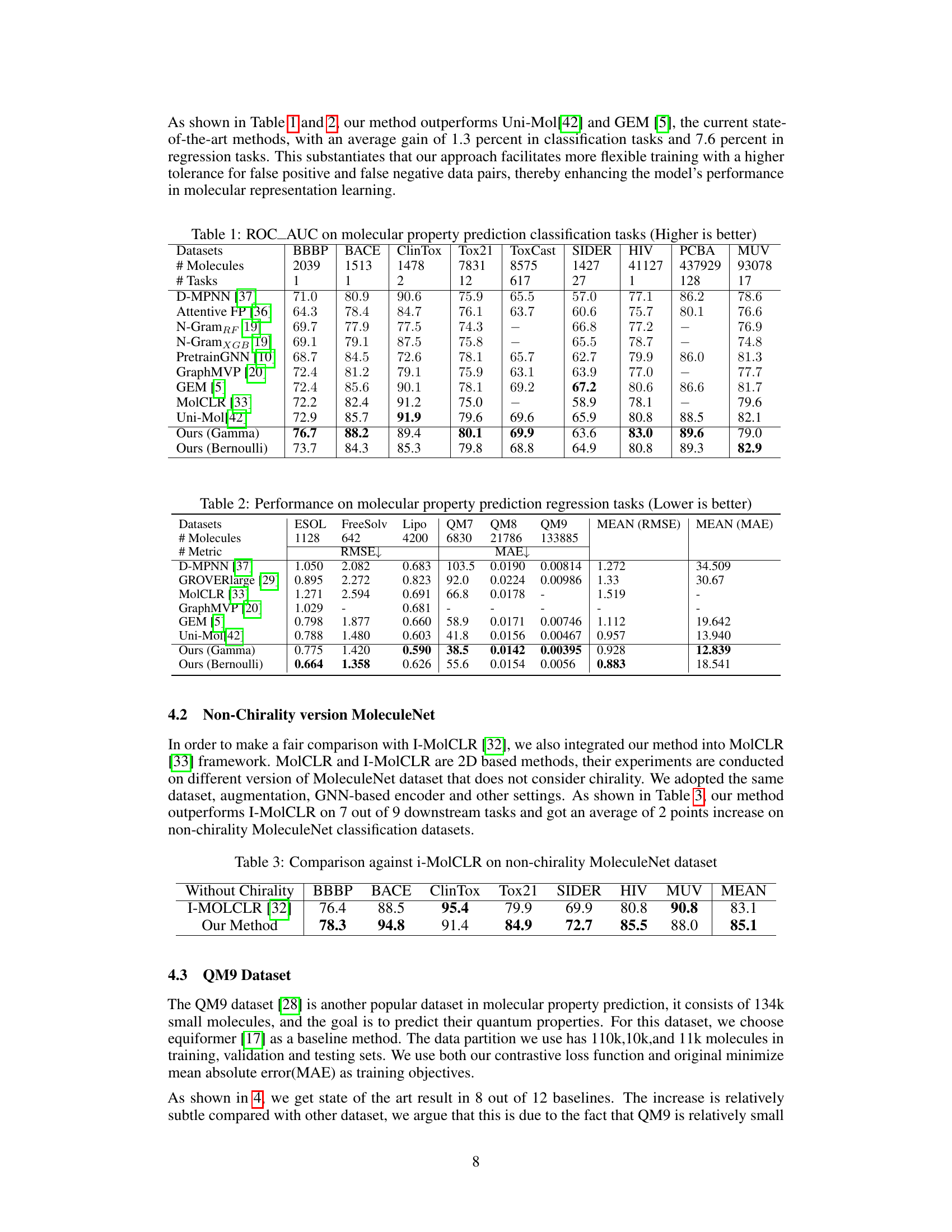
This table presents the results of various molecular property prediction classification tasks on the MoleculeNet dataset. It compares the Area Under the ROC Curve (ROC_AUC) scores achieved by different methods, including several baselines and the proposed method (with Gamma and Bernoulli priors). Higher ROC_AUC scores indicate better performance. The table is organized by dataset and includes the number of molecules and tasks for each dataset.
In-depth insights#
Bayesian MRL#
A hypothetical “Bayesian MRL” framework would represent a significant advancement in molecular representation learning (MRL). Bayesian methods offer a principled way to handle uncertainty inherent in molecular data and model parameters. Instead of relying on point estimates, a Bayesian approach would provide probability distributions over representations, capturing the inherent ambiguity in experimental data and model assumptions. This would lead to more robust and reliable predictions, particularly crucial in drug discovery where uncertainty is unavoidable. A Bayesian framework could naturally incorporate prior knowledge about molecular structures and properties, further improving accuracy and interpretability. Probabilistic graphical models, like Bayesian networks or Markov random fields, could be used to model relationships between molecular features, improving the quality of the learned representations. However, the computational cost of Bayesian methods is often high, representing a key challenge in implementation. Approximations, such as variational inference, would likely be necessary to make the Bayesian MRL approach scalable to large molecular datasets. The development and evaluation of a Bayesian MRL approach would therefore be a substantial contribution to the field, balancing the benefits of probabilistic modeling with the need for computational efficiency.
Probabilistic CL#
The core idea behind Probabilistic Contrastive Learning (CL) is to address the limitations of traditional CL in molecular representation learning, where augmentations can produce unreliable positive and negative pairs. Instead of relying on hard assignments, this approach introduces learnable weights reflecting the probability of a pair’s true label, mitigating the impact of false pairs. A Bayesian framework allows automatic inference of these weights. This probabilistic method is particularly valuable because it adapts to the data’s uncertainty, dynamically adjusting the model. The efficacy of this method lies in its ability to improve the accuracy of learned representations. The Bayesian inference and expectation-maximization optimization process combine to enhance performance significantly. The improved results on multiple benchmarks highlight the method’s generalizability and superior performance compared to standard contrastive learning approaches. The use of learnable weights improves the robustness to noisy data, thereby enabling better feature extraction from molecular data. This novel technique demonstrates an effective and generalizable solution to a pervasive problem in molecular representation learning.
QM9 & MolNet#
The QM9 and MoleculeNet datasets represent cornerstone benchmarks in molecular representation learning. QM9, with its focus on small molecules and readily available quantum chemical properties, enables fine-grained evaluation of model accuracy on specific physicochemical attributes. In contrast, MoleculeNet offers a broader scope, encompassing diverse molecular properties and thus assessing a model’s generalizability across varied chemical spaces. The comparative analysis between results obtained on both datasets provides crucial insights into the robustness and generalizability of the proposed probability-weighted contrastive learning framework. Superior performance on MoleculeNet suggests a model’s capability to extrapolate well to larger, more diverse molecular datasets, while strong results on QM9 highlight the accuracy in predicting specific molecular characteristics. The combined use of both datasets allows for a holistic evaluation of the method’s efficacy, emphasizing its strengths and highlighting areas that could benefit from further development.
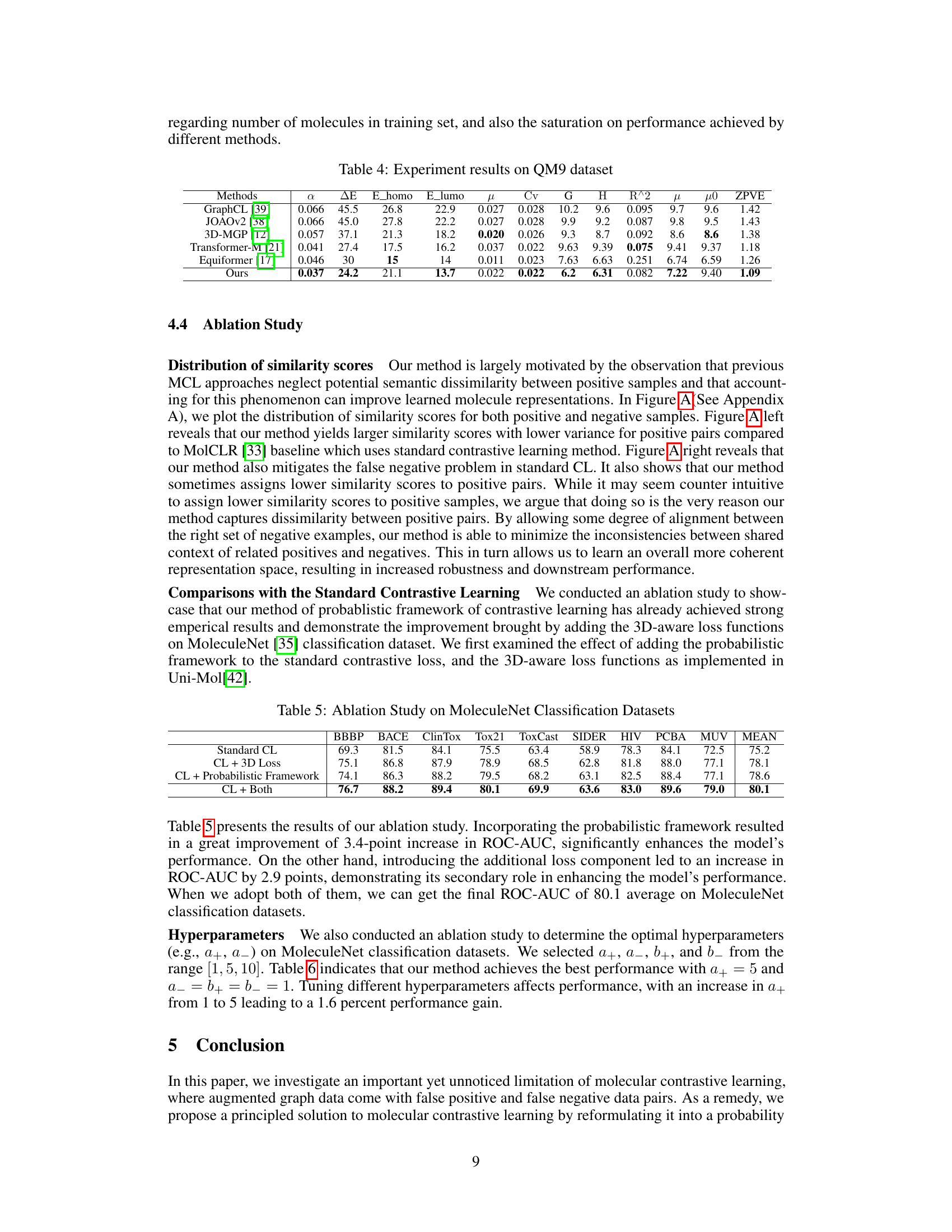
Ablation Study#
The ablation study section in this research paper is crucial for understanding the contribution of each component within the proposed probability-weighted contrastive learning framework. It systematically investigates the impact of removing or altering specific parts of the model to determine their individual effects. The study likely focuses on evaluating the importance of the probabilistic weighting mechanism by comparing results with a standard contrastive loss and various components contributing to the overall accuracy of the molecular representation. The results would reveal whether the addition of probabilistic weights significantly improves the model’s ability to learn robust and accurate representations by mitigating issues caused by false positive/negative pairs, especially from data augmentation. This study’s findings will help assess the model’s efficiency, robustness, and the overall effectiveness of the proposed method compared to existing approaches. Analyzing the ablation study will provide insights into which aspects of the design are most crucial and may highlight potential areas for future improvements or alternative design choices.
Future Work#
Future research directions stemming from this paper could explore several promising avenues. Extending the probabilistic framework to encompass a wider variety of molecular augmentation techniques beyond those tested would enhance the model’s robustness and generalizability. Investigating alternative Bayesian inference methods or incorporating advanced probabilistic models could potentially further improve the accuracy and efficiency of weight estimation. Analyzing the impact of different similarity metrics on the performance of the probabilistic contrastive learning framework is also warranted. Furthermore, a thorough investigation into the sensitivity of the model to hyperparameter choices (particularly those governing the prior distributions) is essential to ensure robust and reliable results across diverse datasets. Finally, applying this probabilistic contrastive learning approach to tasks beyond molecular property prediction, such as drug design or materials science, could unlock its full potential and unveil valuable insights in related fields.
More visual insights#
More on figures

This figure illustrates the molecular contrastive learning framework. (A) shows the overall process: Two stochastic augmentations are applied to each molecule, creating positive pairs. A feature extractor generates representations, and contrastive loss optimizes similarity between positive pairs and dissimilarity between negative pairs. (B), (C), and (D) depict three different feature extractor architectures (Uni-Mol, GCN, and Equiformer) used in different experiments and datasets.
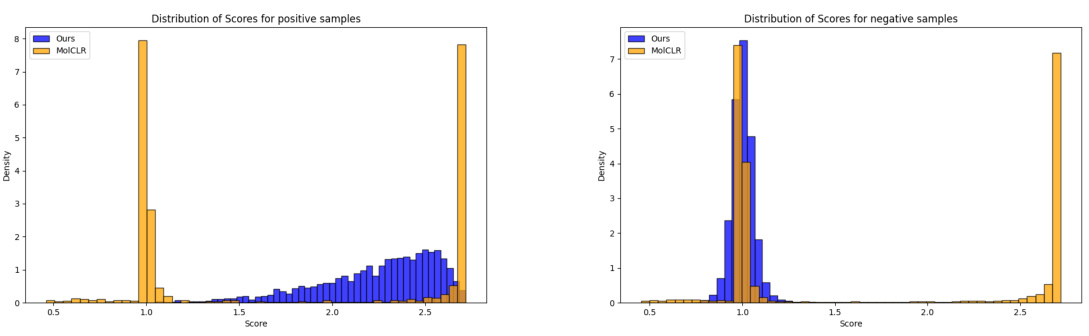
The figure shows the distribution of similarity scores for positive and negative pairs obtained using the proposed method and the baseline MolCLR method. The proposed method demonstrates lower variance in similarity scores for negative pairs, indicating a more focused distribution of negative samples, and higher mean and lower variance for positive pairs, indicating more accurate identification of positive pairs.
More on tables
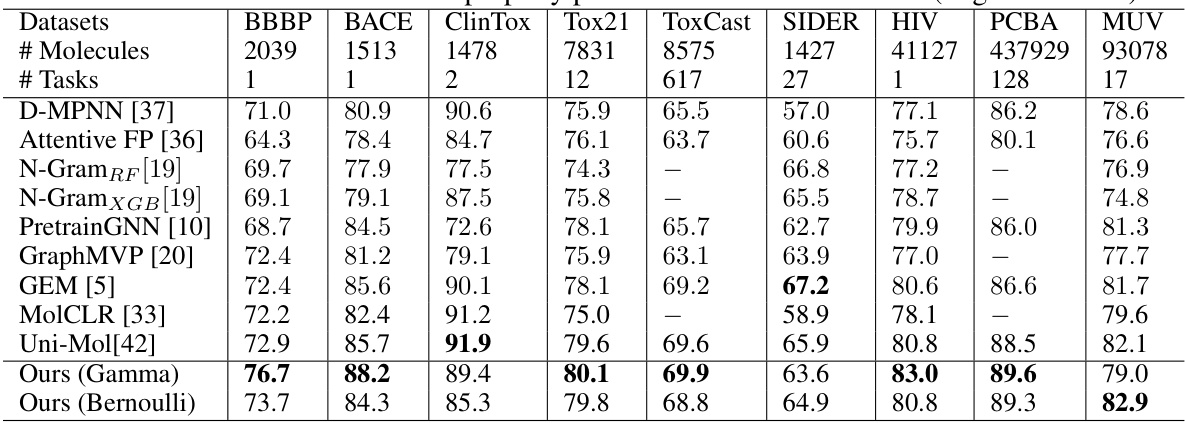
This table presents the performance of various methods on molecular property prediction classification tasks from the MoleculeNet dataset. The ROC-AUC (Receiver Operating Characteristic - Area Under the Curve) score is used as the evaluation metric, with higher scores indicating better performance. The table compares the proposed method (Ours (Gamma) and Ours (Bernoulli)) against several state-of-the-art baselines, including D-MPNN, Attentive FP, N-GramRF, N-GramXGB, PretrainGNN, GraphMVP, GEM, MolCLR, and Uni-Mol. The results are shown for multiple datasets and tasks within the MoleculeNet benchmark, allowing for a comprehensive comparison of the methods.
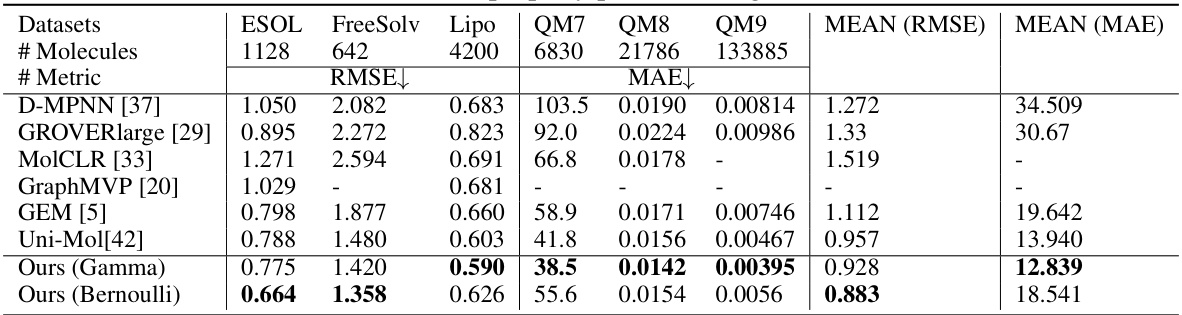
This table presents the results of the proposed method and baseline methods on several molecular property prediction regression tasks from the MoleculeNet dataset. It shows the Root Mean Squared Error (RMSE) and Mean Absolute Error (MAE) for each task and each method. Lower values indicate better performance. The tasks include predicting various physicochemical properties of molecules.

This table presents a comparison of the performance of the proposed method and I-MolCLR on the non-chirality version of the MoleculeNet dataset. The table shows the ROC-AUC scores for both methods across nine different molecular property prediction tasks. The results demonstrate the superiority of the proposed method over the baseline.
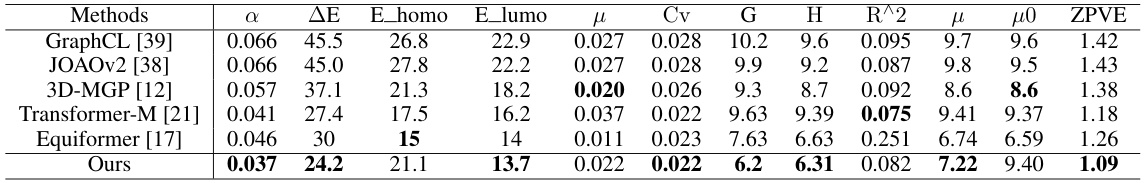
This table presents the results of the experiments conducted on the QM9 dataset. It compares the performance of the proposed method against several state-of-the-art baselines across 12 different molecular property prediction tasks. The properties are represented by different symbols, such as α (dipole polarizability), ΔE (atomization energy), E_homo (highest occupied molecular orbital energy), E_lumo (lowest unoccupied molecular orbital energy), μ (dipole moment), Cv (heat capacity), G (Gibbs free energy), H (enthalpy), R^2 (coefficient of determination), μ (dipole moment), μ0 (isotropic magnetic shielding), and ZPVE (zero-point vibrational energy). The performance is measured by the mean absolute error (MAE).
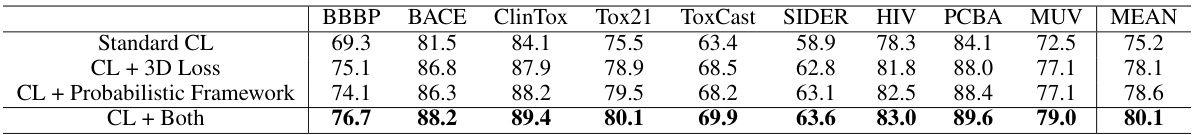
This table presents the ablation study results on the MoleculeNet classification datasets. It shows the performance of four different model configurations: using only standard contrastive learning (CL); CL with the addition of 3D loss; CL with the addition of the proposed probabilistic framework; and CL with both 3D loss and the probabilistic framework. The results show a significant improvement in performance when adding both the probabilistic framework and 3D loss.
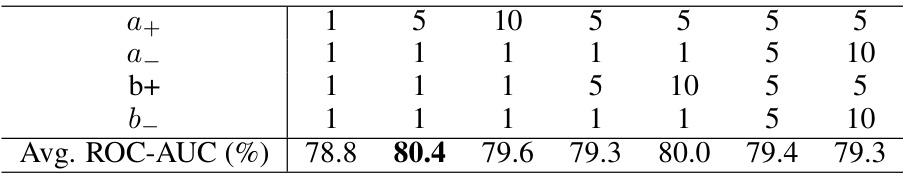
This table presents the results of an ablation study on the hyperparameters used in the MoleculeNet classification experiments. It shows how different values of (a_+), (a_-), (b_+), and (b_-) affect the average ROC-AUC score. The best performance (80.4%) is achieved with (a_+ = 5), (a_- = 1), (b_+ = 1), and (b_- = 1).

This table shows the range of hyperparameters explored during the experiments on the MoleculeNet dataset. It is broken down by dataset size (Small, Large, HIV) and lists the ranges considered for learning rate, batch size, number of epochs, pooler dropout rate, and warmup ratio.

This table presents a comparison of the proposed method’s performance against the i-MolCLR method on the non-chirality version of the MoleculeNet dataset. It shows the ROC-AUC scores for both methods across nine different molecular property prediction tasks. The results demonstrate that the proposed method outperforms i-MolCLR in most tasks, indicating its effectiveness in handling molecular data with potential false positive and false negative pairs.

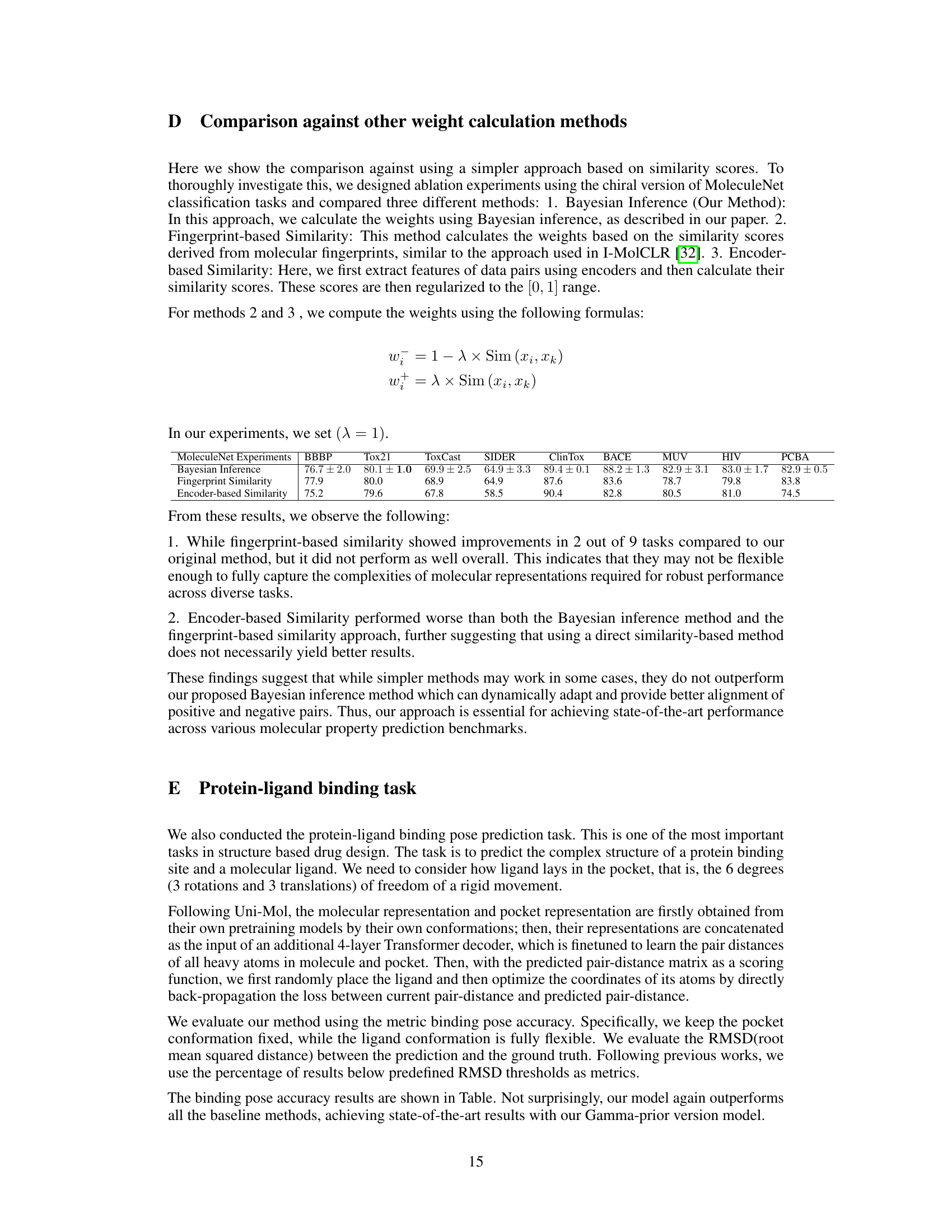
This table presents the performance of different methods on a protein-ligand binding pose prediction task. The task involves predicting the 3D structure of a protein-ligand complex. The performance is measured by the Root Mean Squared Deviation (RMSD) between the predicted and ground truth structures. Lower RMSD values indicate better performance. The table shows the percentage of results with RMSD values below different thresholds (1.0 Å, 1.5 Å, 2.0 Å, 3.0 Å, and 5.0 Å). The results demonstrate that the proposed method (Ours) outperforms existing state-of-the-art methods.

This table presents the results of an ablation study conducted to evaluate the impact of different hyperparameter settings on the performance of the proposed model. Specifically, it examines the effect of varying the shape parameters au and bu of the Gamma distribution used to model the positive weights wi+. The table shows that the best performance is achieved with au = bu = 5, with minimal differences observed for other settings, suggesting a degree of robustness to these specific hyperparameters.
Full paper#