↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Predicting the effects of protein mutations is critical for various applications, but existing methods struggle to extract relevant local structural information. Current approaches often rely on global protein representations which may overlook crucial local micro-environmental details that significantly impact mutation effects. This limitation hinders their predictive accuracy and robustness.
This research introduces a novel retrieval-augmented framework that addresses this by creating a database of local structure motif embeddings from a pre-trained protein structure encoder. This allows for efficient retrieval of similar local structure motifs during mutation effect prediction, effectively capturing crucial co-evolutionary information. The new method significantly enhances accuracy, achieving state-of-the-art results on multiple benchmark datasets, offering a scalable solution for studying mutation effects.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in protein engineering and computational biology. It presents a novel, scalable solution for mutation effect prediction, significantly improving accuracy. This opens new avenues for drug design, biofuel production, and understanding genetic diseases. The innovative retrieval-augmented framework and superior performance on benchmark datasets establish its value for the field.
Visual Insights#
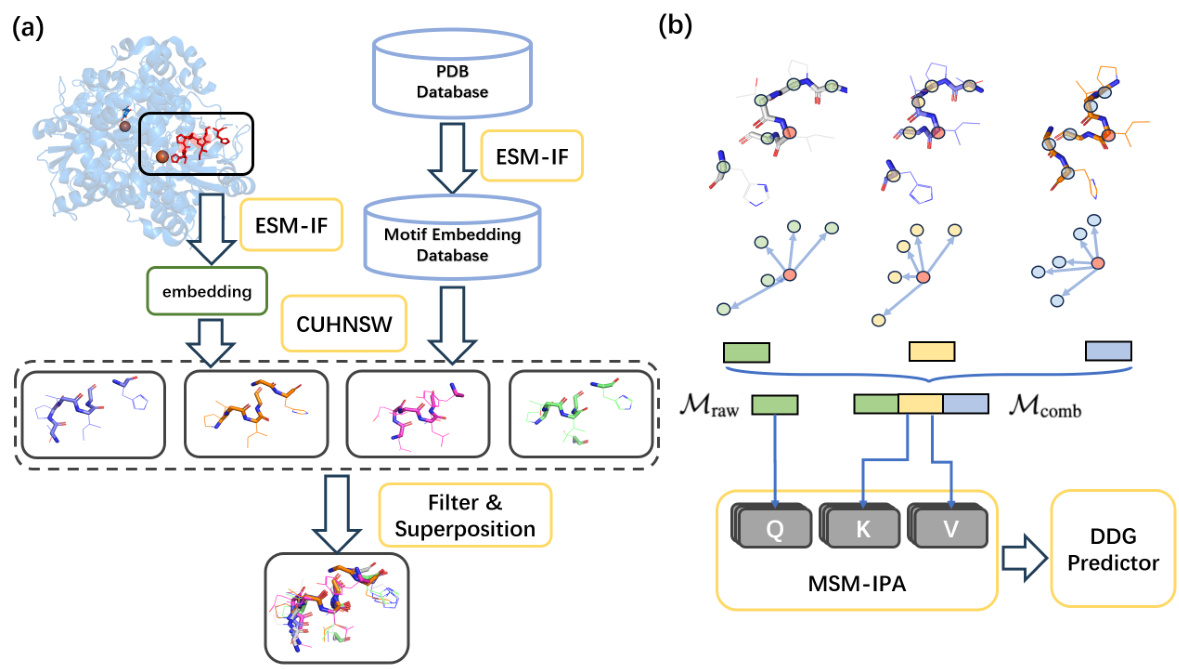
This figure illustrates the proposed retrieval-augmented framework for protein mutation effect prediction. Panel (a) shows the Multiple Structure Motif Alignment (MSMA) process, which involves extracting local structure motifs from a protein structure using ESM-IF, storing them in a database (SMEDB), and retrieving similar motifs using CUHNSW. Panel (b) shows the MSM-Mut model architecture, which incorporates the retrieved motifs to predict mutation effects using a Multi-Structure Motif Invariant Point Attention (MSM-IPA) module.
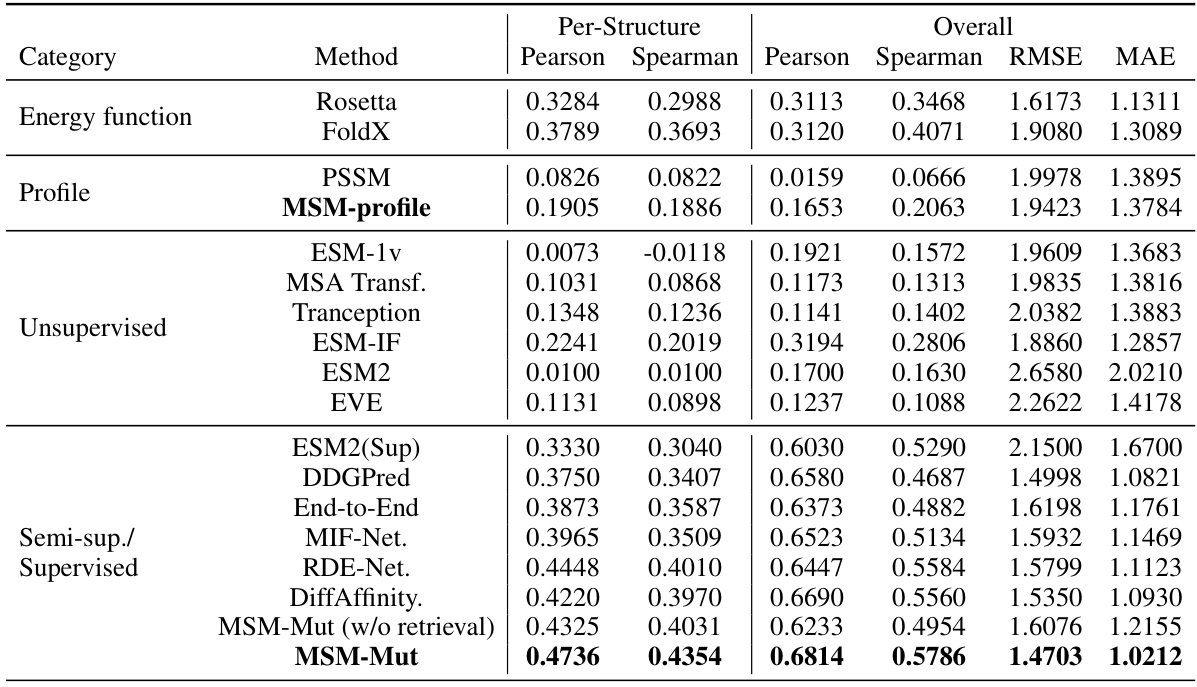
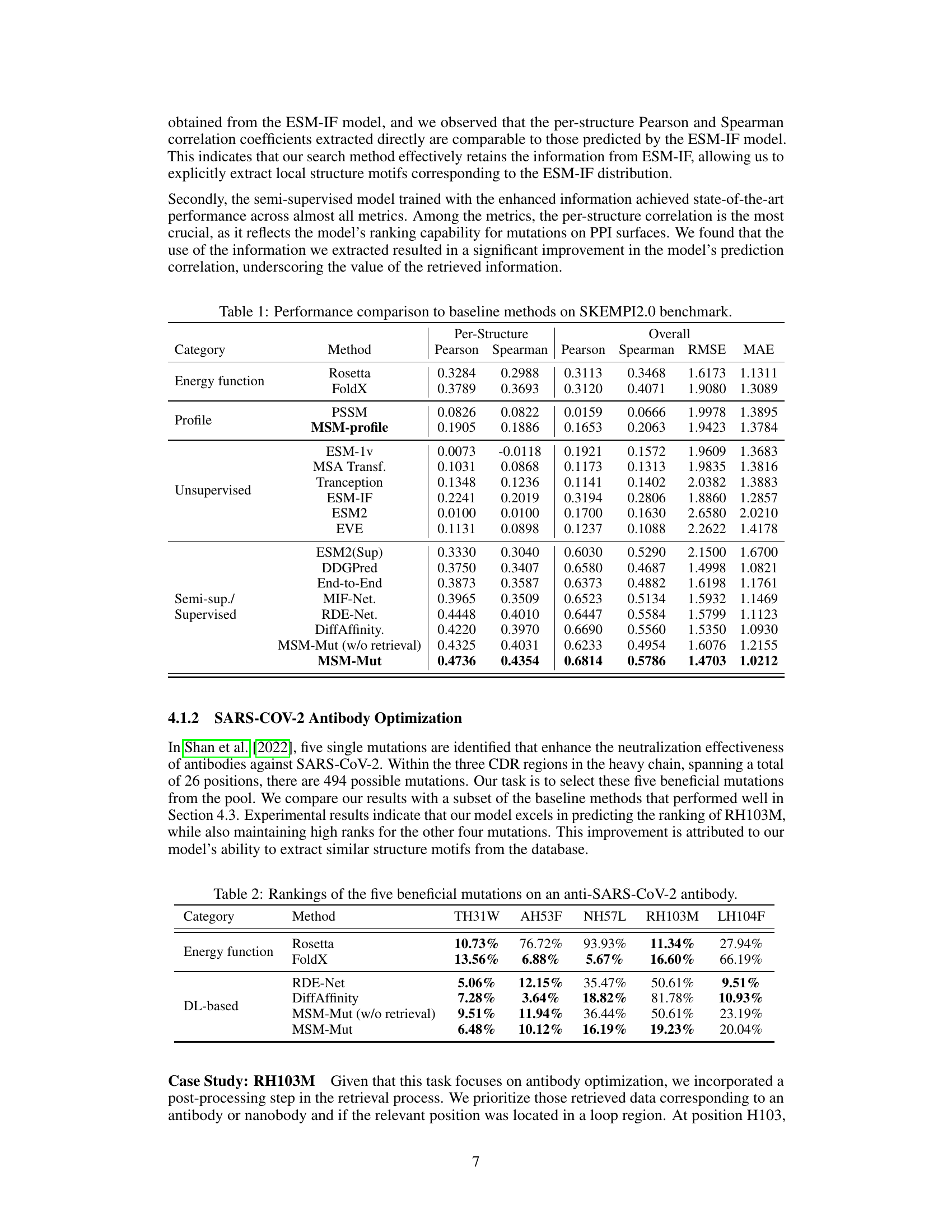
This table compares the performance of the proposed MSM-Mut model against various baseline methods on the SKEMPI2.0 benchmark dataset for mutation effect prediction on protein-protein interfaces. The metrics used include Pearson and Spearman correlation coefficients for per-structure and overall performance, as well as RMSE and MAE for evaluating the accuracy of predicted binding energy changes. The baseline methods represent a range of approaches, including energy function-based methods (Rosetta, FoldX), profile-based methods (PSSM, MSM-profile), unsupervised learning methods (ESM-1v, MSA Transformer, Tranception, ESM-IF, ESM2, EVE), and supervised learning methods (ESM2(Sup), DDGPred, End-to-End, MIF-Net, RDE-Net, DiffAffinity). The table highlights the superior performance of the MSM-Mut model, particularly in terms of per-structure correlation metrics.
In-depth insights#
Local Motif Focus#
A ‘Local Motif Focus’ in protein mutation effect prediction offers significant advantages over global sequence or structure-based methods. By concentrating on the microenvironment surrounding a mutation site, it allows for a more refined understanding of how local structural changes impact protein function. This approach is particularly valuable in protein engineering, where subtle modifications to specific motifs can have dramatic effects. Focusing on local motifs allows for more efficient data retrieval and reduced computational demands compared to analyzing entire protein structures. The use of pre-trained structural encoders to generate embeddings of local motifs further enhances the accuracy and efficiency of this approach, enabling the identification of functionally important regions, even across distantly related proteins. Leveraging a database of local motif embeddings allows for the rapid retrieval of similar structural contexts, which provides crucial auxiliary information for predicting the effects of mutations. This ultimately facilitates more accurate and robust predictions, leading to advancements in fields such as drug discovery and genetic disease research.
Retrieval Augment#
Retrieval augmentation, in the context of protein mutation effect prediction, is a powerful technique that significantly enhances predictive accuracy. It leverages the wealth of information present in protein structure databases by retrieving similar local structure motifs for a given mutation. This approach moves beyond relying solely on global sequence or structure comparisons, which often fail to capture the crucial micro-environmental effects of point mutations. The use of a pre-trained protein structure encoder to generate embeddings of local motifs is a key innovation, allowing for efficient similarity searches within a vast database. This process is computationally efficient, making it scalable for large-scale studies. Furthermore, the combination of retrieval-based information with traditional methods like Multiple Sequence Alignments often yields superior performance, demonstrating the complementary nature of these approaches. The successful application of retrieval augmentation highlights the importance of integrating diverse data sources and focusing on local structural context for accurate prediction of protein mutation effects.
MSM-IPA Model#
The MSM-IPA (Multi-Structure Motif Invariant Point Attention) model represents a novel architecture designed for protein mutation effect prediction. It leverages a retrieval-augmented framework, efficiently incorporating similar local structure information from a pre-trained protein structure encoder. This contrasts with traditional methods relying solely on global sequence or structure alignments. The model’s core innovation lies in its ability to aggregate information from multiple retrieved, locally similar structure motifs. This is achieved through an attention mechanism that is invariant to overall rotation and translation, focusing on the crucial local biochemical microenvironment surrounding the mutation site. The integration of MSM-IPA with a structure motif database (SMEDB) offers a scalable and robust approach, surpassing the state-of-the-art in performance across multiple benchmark datasets. This improvement stems from the model’s ability to capture complementary coevolutionary information compared to traditional methods, thus improving predictive accuracy and robustness.
Benchmark Results#
A dedicated section detailing benchmark results is crucial for evaluating the performance of a proposed method. It should present a comparison against state-of-the-art baselines across multiple datasets, using relevant metrics. The choice of metrics is vital: selecting those that directly address the problem’s core aspects is key. Presenting results with error bars or confidence intervals highlights the reliability and statistical significance of the findings. Visualizations like bar charts or tables aid in clear, concise comparisons. It’s also crucial to discuss any limitations or caveats in the benchmarking process, such as dataset biases or limitations of the baselines, providing a nuanced perspective and fostering trust in the presented results. Highlighting the improvements and providing context regarding the significance of the performance gains adds to the section’s value. A comprehensive analysis of benchmark results builds confidence in the proposed method’s robustness and effectiveness.
Future Directions#
Future research could explore expanding the local structure motif database to encompass a wider range of protein structures and mutations, potentially enhancing predictive accuracy. Improving the efficiency of the retrieval mechanism is another key area, as faster retrieval would enable analysis of larger datasets and more complex mutations. Investigating the potential of integrating other data sources, such as evolutionary information or experimental data, into the framework could provide a more holistic view of mutation effects. Finally, exploring applications to specific biological problems, such as drug design and disease modeling, is crucial to demonstrate the practical value of this approach.
More visual insights#
More on figures
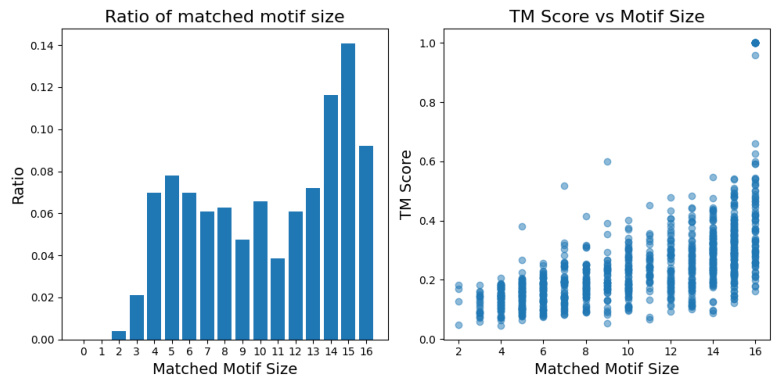
This figure shows two plots. The left plot is a histogram showing the distribution of the number of matched motifs of different sizes retrieved by the CUHNSW algorithm combined with ESM-IF embeddings. The right plot shows the relationship between TM-score (a measure of structural similarity) and the number of matched motifs. The plots demonstrate that the method effectively retrieves local structural motifs even from structurally unrelated proteins, highlighting the capability of the approach to capture analogous local structural information.

This figure shows two highly similar local antibody structures retrieved by the MSM (Multiple Structure Motif) alignment method. The structures, from proteins 5KOV and 7FAE, highlight that similar local structural motifs can be found in different proteins, even when the overall protein structures differ significantly. This demonstrates the power of the MSM method to identify functionally relevant local structures.

This figure visualizes the results of Multiple Structure Motif Alignment (MSMA), a key component of the MSM-Mut model. It shows two sets of highly similar local antibody structures that were retrieved using the method. The structures, despite being from different proteins (5KOV and 7FAE), share highly similar local micro-environments around the central amino acid. This highlights the ability of the MSMA to find structurally similar regions even when the overall global sequence/structure similarity is low. The similarity in local structure motifs supports the model’s approach to leveraging these local features for accurate mutation effect prediction.
More on tables
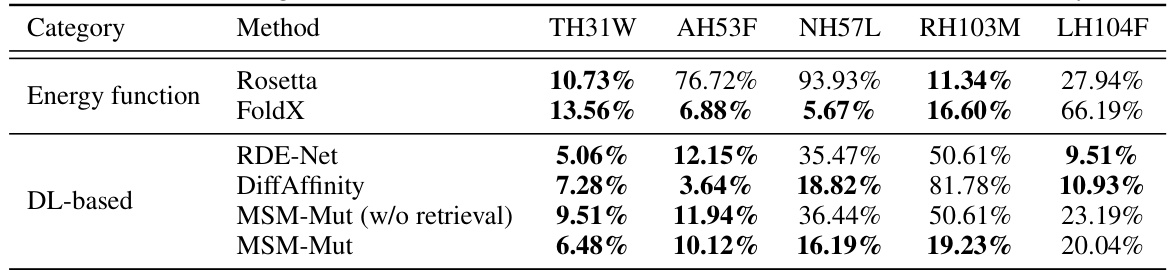
This table compares the ranking of five beneficial mutations (TH31W, AH53F, NH57L, RH103M, LH104F) identified by Shan et al. [2022] as enhancing the neutralization effectiveness of antibodies against SARS-CoV-2, as predicted by different methods including Rosetta, FoldX, RDE-Net, DiffAffinity, and the proposed MSM-Mut (with and without retrieval). It highlights the relative performance of each method in predicting the rank order of these beneficial mutations.
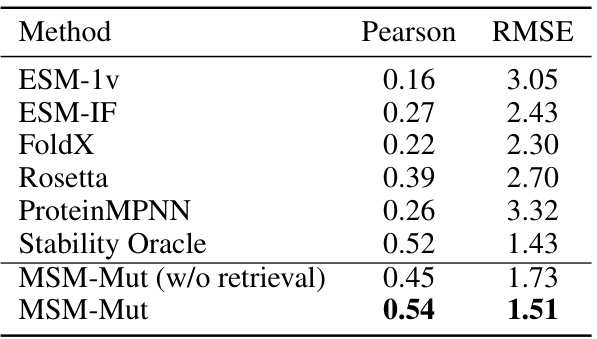
This table compares the performance of the MSM-Mut model against various baseline methods on two datasets: S669 and Novozymes. The Pearson correlation and RMSE (Root Mean Square Error) are reported for each method on each dataset. The S669 dataset contains protein variants not found in commonly used training sets, providing a robust evaluation. The Novozymes dataset is a novel enzyme thermostability dataset with limited sequence similarity to existing data, testing generalization capabilities. The results demonstrate the superior performance of MSM-Mut, especially on the more challenging Novozymes dataset.
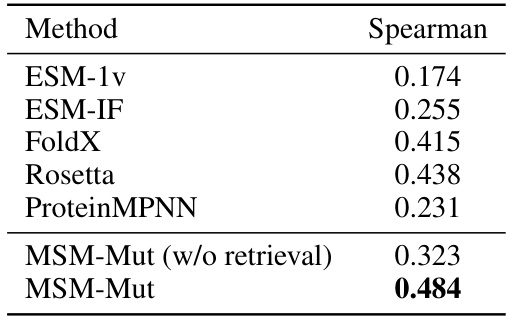
This table compares the performance of the proposed MSM-Mut model against several baseline methods on two datasets: S669 and Novozymes. The Pearson and RMSE (Root Mean Squared Error) metrics are used to evaluate the model’s performance in predicting the stability changes of proteins. The table highlights the superior performance of MSM-Mut, especially when comparing Spearman correlation.
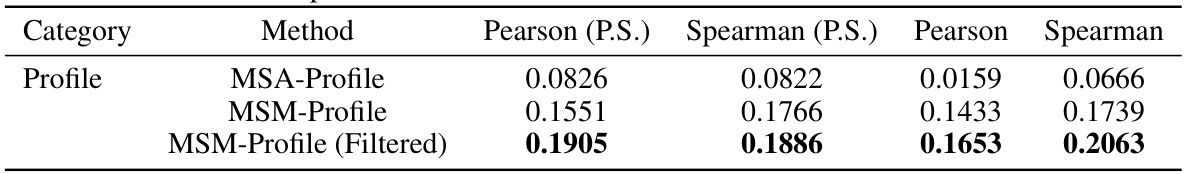
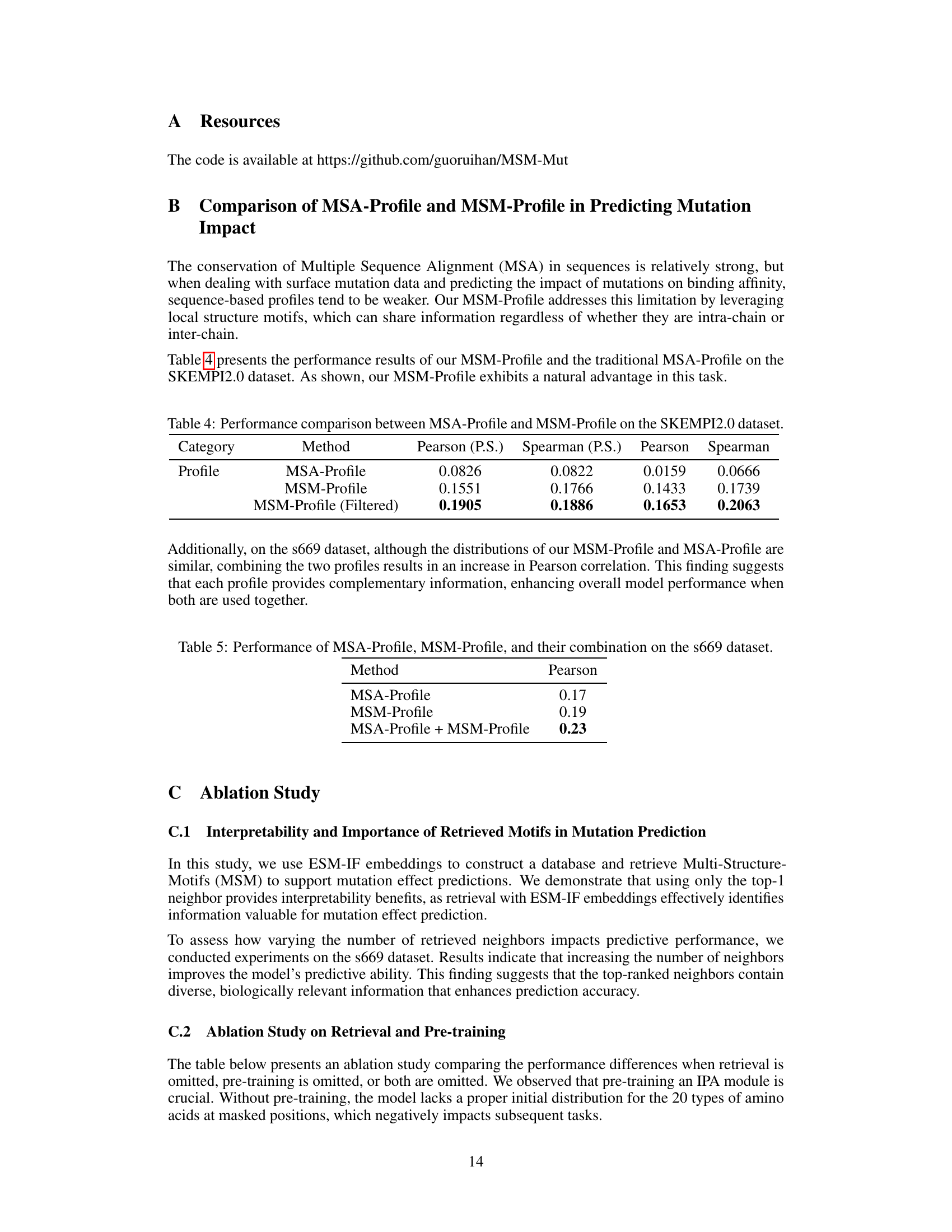
This table compares the performance of MSA-Profile and MSM-Profile on the SKEMPI2.0 dataset. It shows the Pearson and Spearman correlation coefficients for both profiles, demonstrating the superior performance of MSM-Profile in predicting mutation effects on protein-protein interactions. The results highlight that incorporating local structure information, as done by MSM-Profile, leads to significantly improved predictive accuracy compared to traditional MSA-based approaches.

This table compares the performance of MSA-Profile, MSM-Profile, and their combination on the s669 dataset. It shows that combining both profiles leads to improved performance, suggesting that each profile provides complementary information.
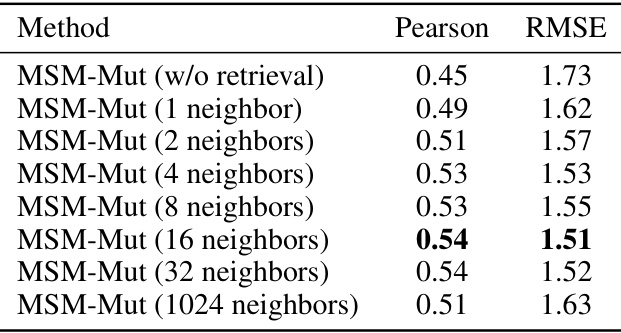
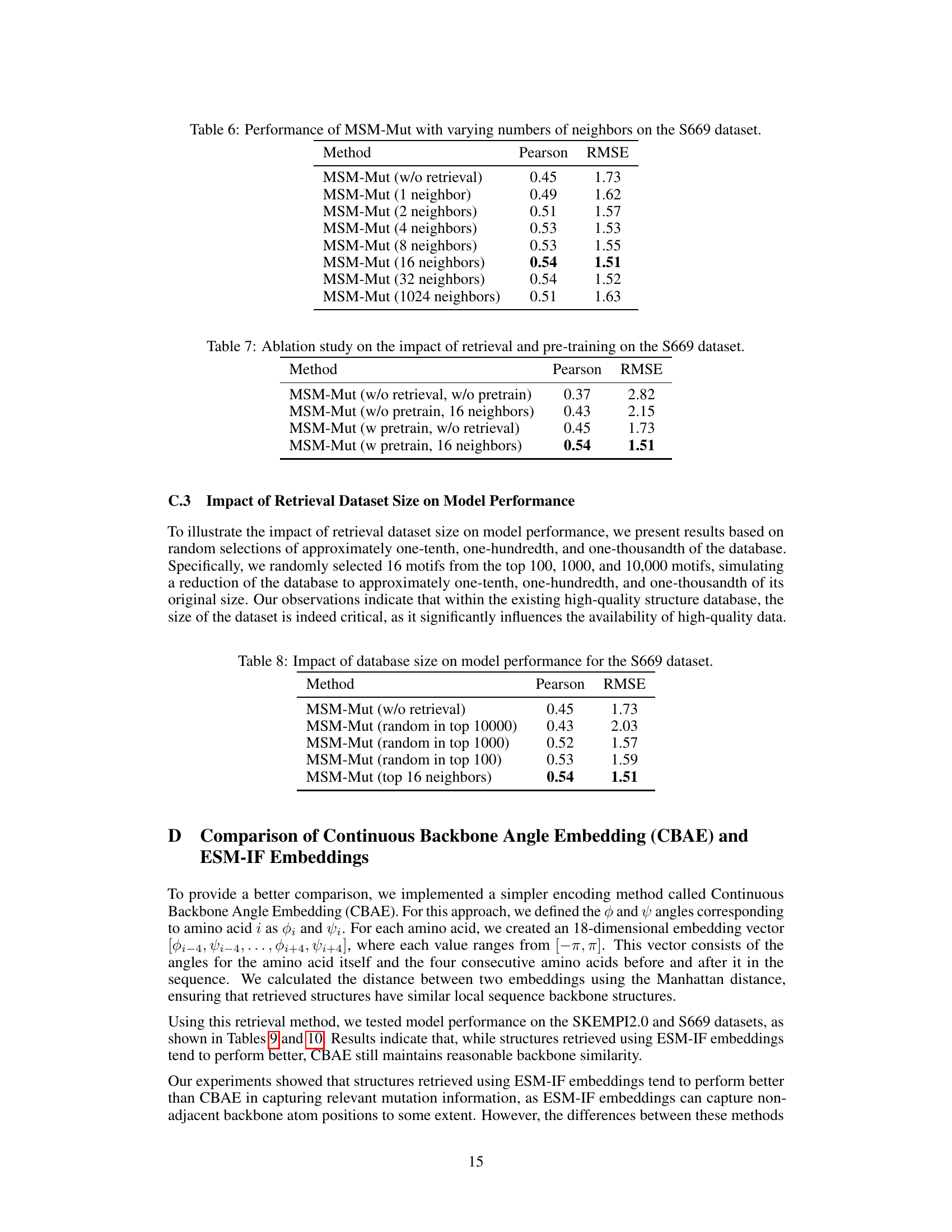
This table presents the results of an ablation study that investigates the impact of varying the number of retrieved neighbors on the performance of the MSM-Mut model. The study was conducted on the S669 dataset, a widely used benchmark for protein stability prediction. The table shows that increasing the number of neighbors from 1 to 16 improves the model’s performance (as measured by Pearson correlation and RMSE). However, increasing the number of neighbors beyond 16 does not lead to further improvement, and even results in a slight decrease in performance when using 1024 neighbors.

This ablation study shows the impact of removing the retrieval mechanism and/or the pre-training step on the performance of the MSM-Mut model, using Pearson correlation and RMSE as evaluation metrics on the S669 dataset. The results highlight the importance of both retrieval and pre-training for achieving optimal performance.

This table shows the performance of the MSM-Mut model on the S669 dataset when using different sizes of the retrieved structure motif database. The results demonstrate that the performance of the MSM-Mut model is influenced by the size of the database, with larger databases leading to better performance. The table compares performance using random selections of 100, 1000, and 10000 motifs and the top 16 neighbors, against the model without retrieval.
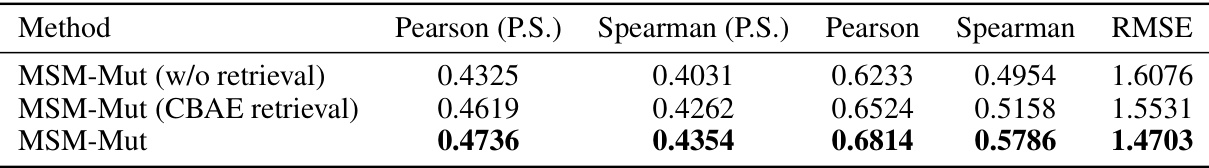
This table compares the performance of the MSM-Mut model with and without using Continuous Backbone Angle Embedding (CBAE) for retrieval on the SKEMPI2.0 benchmark dataset. It shows the Pearson and Spearman correlation coefficients for per-structure and overall metrics, and also includes the RMSE (root mean squared error). The results indicate that using ESM-IF embeddings for retrieval leads to better performance than CBAE on this dataset.

This table presents the performance comparison results of MSM-Mut with and without CBAE retrieval on the S669 dataset. It shows the Pearson correlation and RMSE values for each method. The comparison highlights the impact of using Continuous Backbone Angle Embedding (CBAE) for retrieval on the model’s predictive accuracy.
Full paper#