↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Predicting relationships between biological entities (like drug-target-protein) is crucial for drug discovery and personalized medicine. Current deep learning methods focus on individual entities, neglecting the valuable information within association patterns among them. This leads to inaccurate predictions and limited understanding of underlying biological mechanisms.
The proposed method, Pattern-BERP, directly addresses this issue. It uses a novel fusion method that incorporates association patterns, improving predictive accuracy by 4-23%. A bind-relation module enhances the representation of low-order associations. The detailed analysis of association patterns enhances the model’s interpretability, thus revealing crucial biological mechanisms. This makes the model not only accurate but also helps researchers understand the underlying biology better, making it more applicable in real-world scenarios.
Key Takeaways#
Why does it matter?#
This paper is crucial because it significantly advances biological entity relationship prediction. Its novel association pattern-aware fusion method improves accuracy by 4-23% compared to existing methods, opening exciting avenues for drug discovery and personalized medicine. The detailed explanation of association patterns enhances interpretability, which is vital for real-world applications.
Visual Insights#
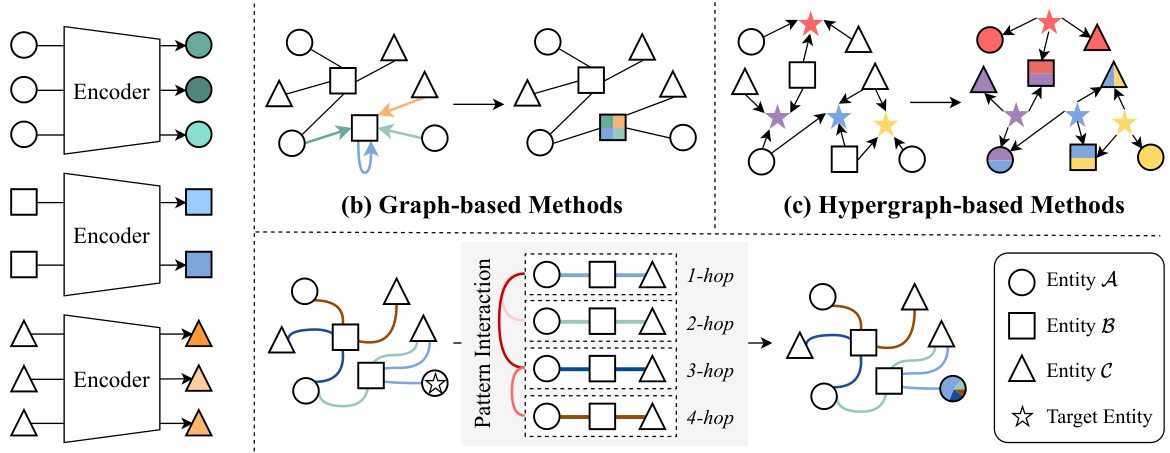
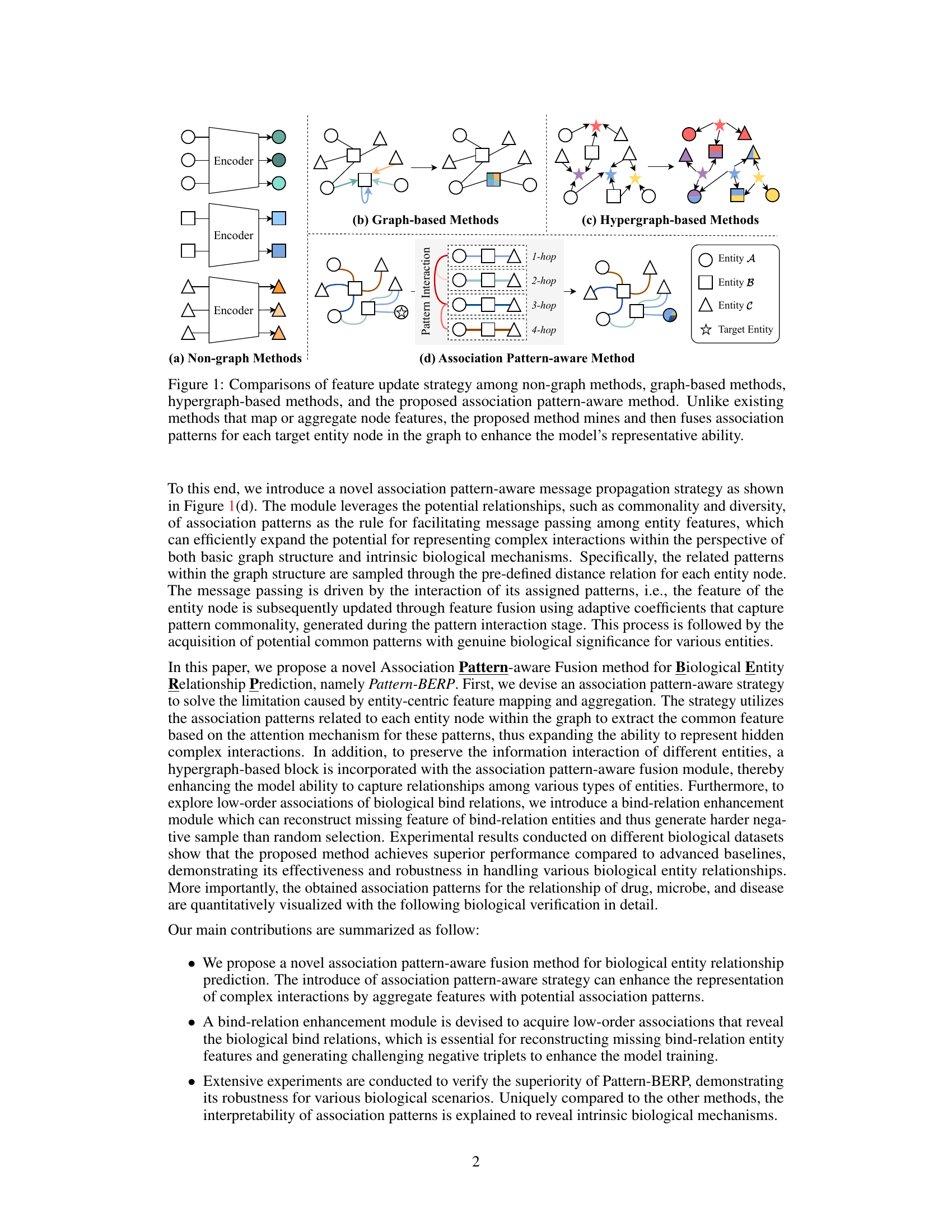
This figure compares four different approaches for updating features in a graph neural network. Non-graph methods concatenate independently encoded features. Graph-based methods use graph structure for feature propagation. Hypergraph-based methods utilize hypergraph structure and complex feature aggregation. The proposed association pattern-aware method is distinct in that it identifies and integrates association patterns to improve the representation of target entities.
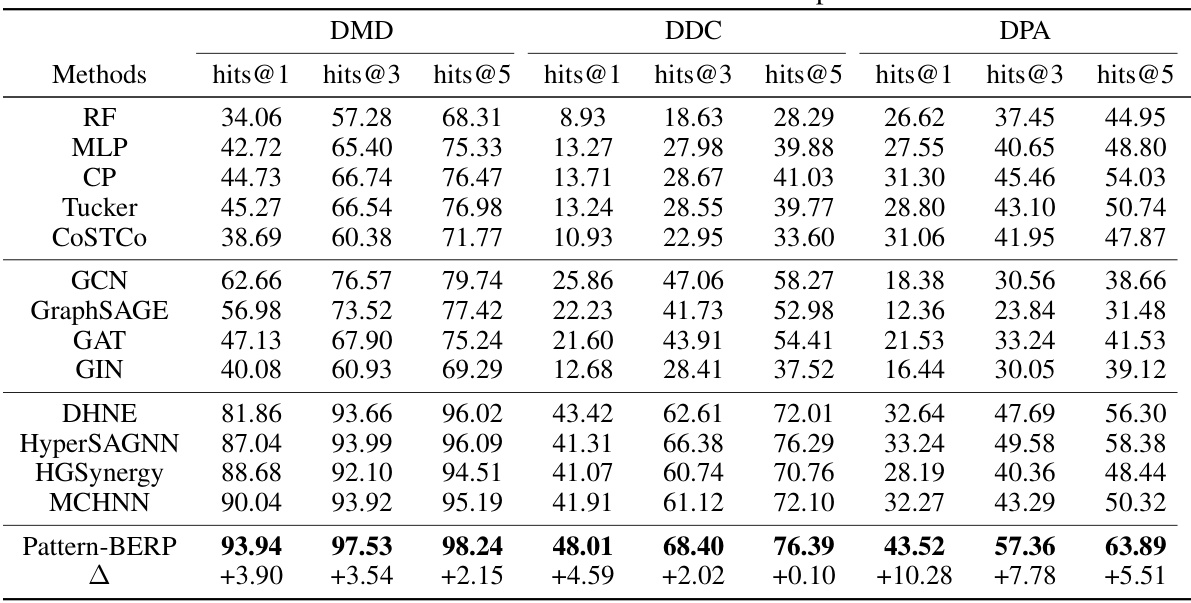
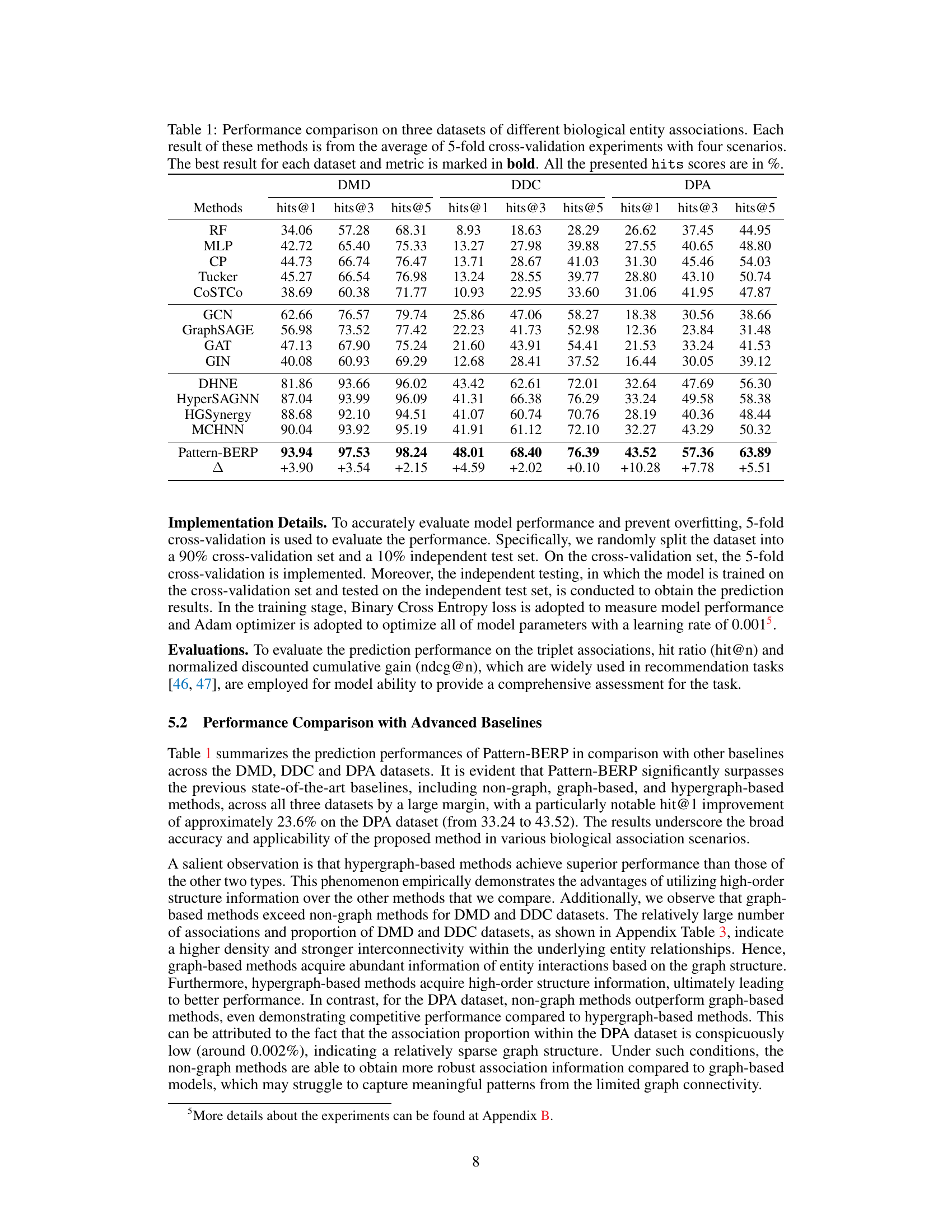
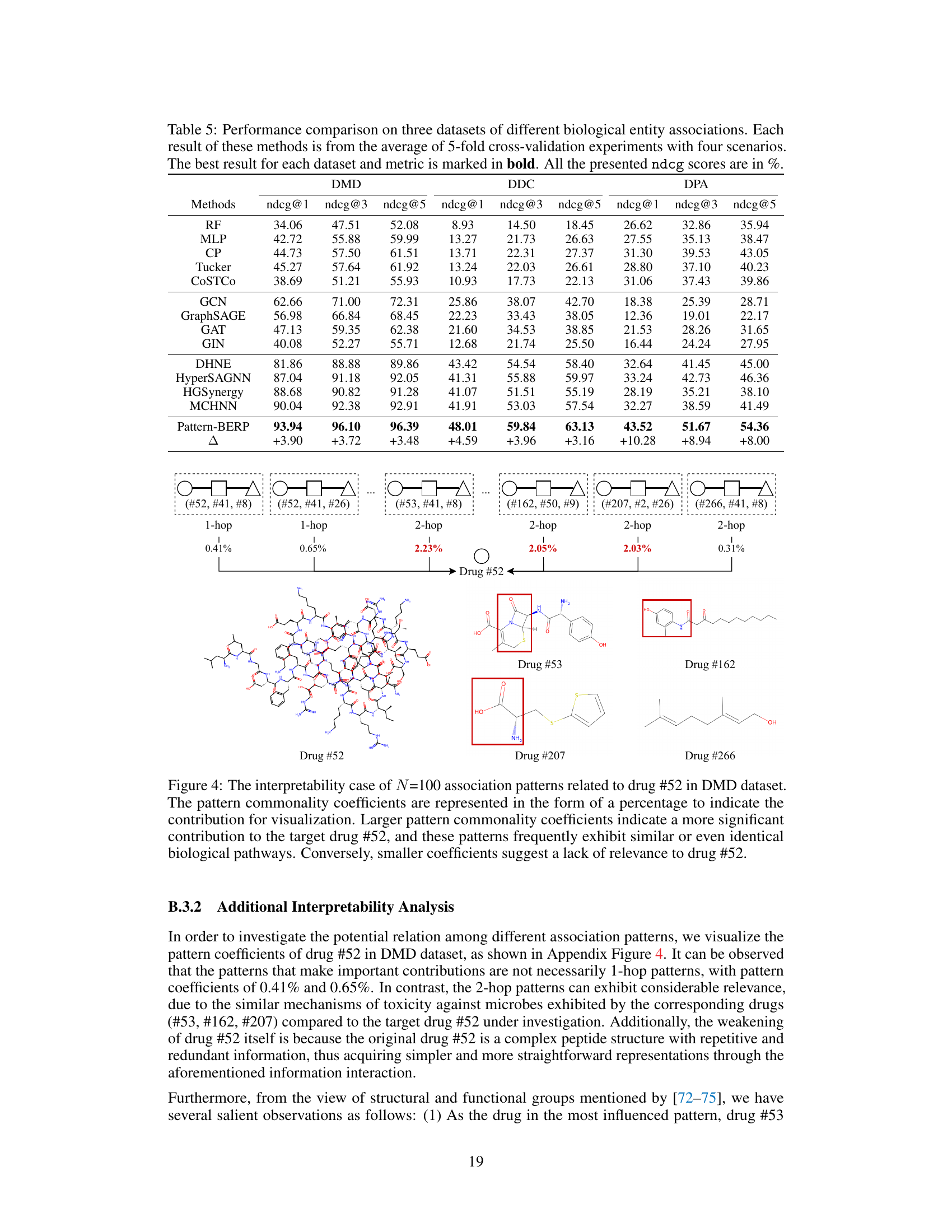
This table presents a comparison of the proposed Pattern-BERP method against several baseline methods for biological entity relationship prediction. The performance is evaluated using hit@1, hit@3, and hit@5 metrics across three different datasets (DMD, DDC, DPA), representing various types of biological associations. The results are averaged over five-fold cross-validation, with the best results for each dataset and metric highlighted in bold. The table provides a quantitative comparison demonstrating Pattern-BERP’s superior performance.
In-depth insights#
Pattern Fusion#
The concept of ‘Pattern Fusion’ in a research paper likely refers to a method that integrates and analyzes recurring patterns within data to improve the overall model’s performance or understanding. This might involve identifying significant motifs or substructures from different sources and combining their representations. The effectiveness of pattern fusion hinges on several key factors. First, the chosen patterns should be relevant and informative, reflecting underlying relationships or causal structures. Second, the fusion method itself must be appropriate; simple concatenation may be insufficient, and more sophisticated techniques like attention mechanisms or graph neural networks could be necessary for handling complex interactions. Successful pattern fusion requires careful feature engineering and selection. The process needs to capture the essence of the patterns while avoiding redundancy or noise, and dimensionality reduction may be crucial for managing high-dimensional data. The interpretability of the fused representations is also important, as understanding how the patterns contribute to model output enhances transparency and trust. Ultimately, the value of pattern fusion lies in its potential to improve accuracy, generalization, and especially the insights derived from the analysis. By revealing latent structures and relationships, it may lead to novel discoveries and improved decision-making in applications such as drug discovery or disease prediction.
Bind Relation#
The concept of “Bind Relation” in the context of biological entity relationship prediction is crucial for capturing complex interactions between entities that are not directly linked. A bind relation module addresses the limitation of existing methods that primarily focus on higher-order relationships by explicitly modeling low-order associations. This is particularly important because low-order interactions often represent fundamental biological mechanisms that are missed by solely focusing on high-order paths. The module’s core function is to reconstruct potentially missing information about bind relations, effectively enriching the entity representations with crucial contextual details. Furthermore, by generating hard negative samples, the bind relation module enhances the model’s ability to distinguish true from false relationships, improving its overall predictive performance. Interpretability is another key feature, allowing analysis to demonstrate how these bind relations reflect underlying biological processes and contribute to the model’s success.
Interpretability#
The concept of interpretability in machine learning models, especially within the context of biological entity relationship prediction, is crucial for establishing trust and facilitating scientific discovery. Interpretable models allow researchers to understand the underlying reasoning behind predictions, moving beyond simple accuracy metrics. In the provided research paper, interpretability is approached by visualizing association patterns and their contributions to predictions. By examining the strength of these patterns (represented as commonality coefficients), researchers gain insight into the biological mechanisms driving the relationships, potentially revealing novel pathways and interactions. The focus is on how different patterns contribute to specific entity relationships. High commonality coefficients indicate biologically relevant and significant patterns, supporting the model’s capacity for discovering meaningful insights. However, the level of interpretability hinges on the sophistication of the visualization and the extent to which the identified patterns align with established biological knowledge. Further research could explore advanced visualization techniques and integrate external biological databases to enhance interpretability and facilitate the translation of model findings into concrete biological hypotheses.
Ablation Study#
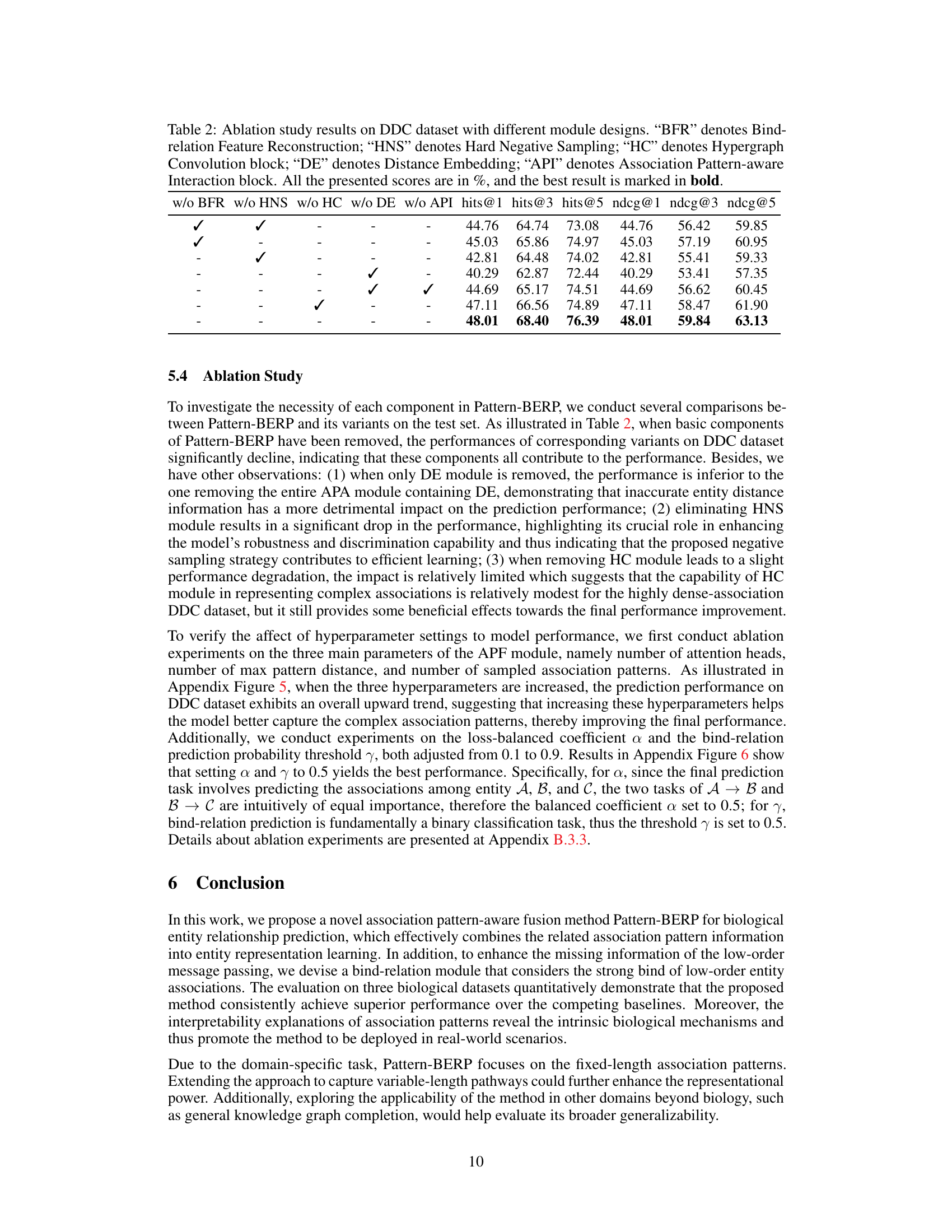
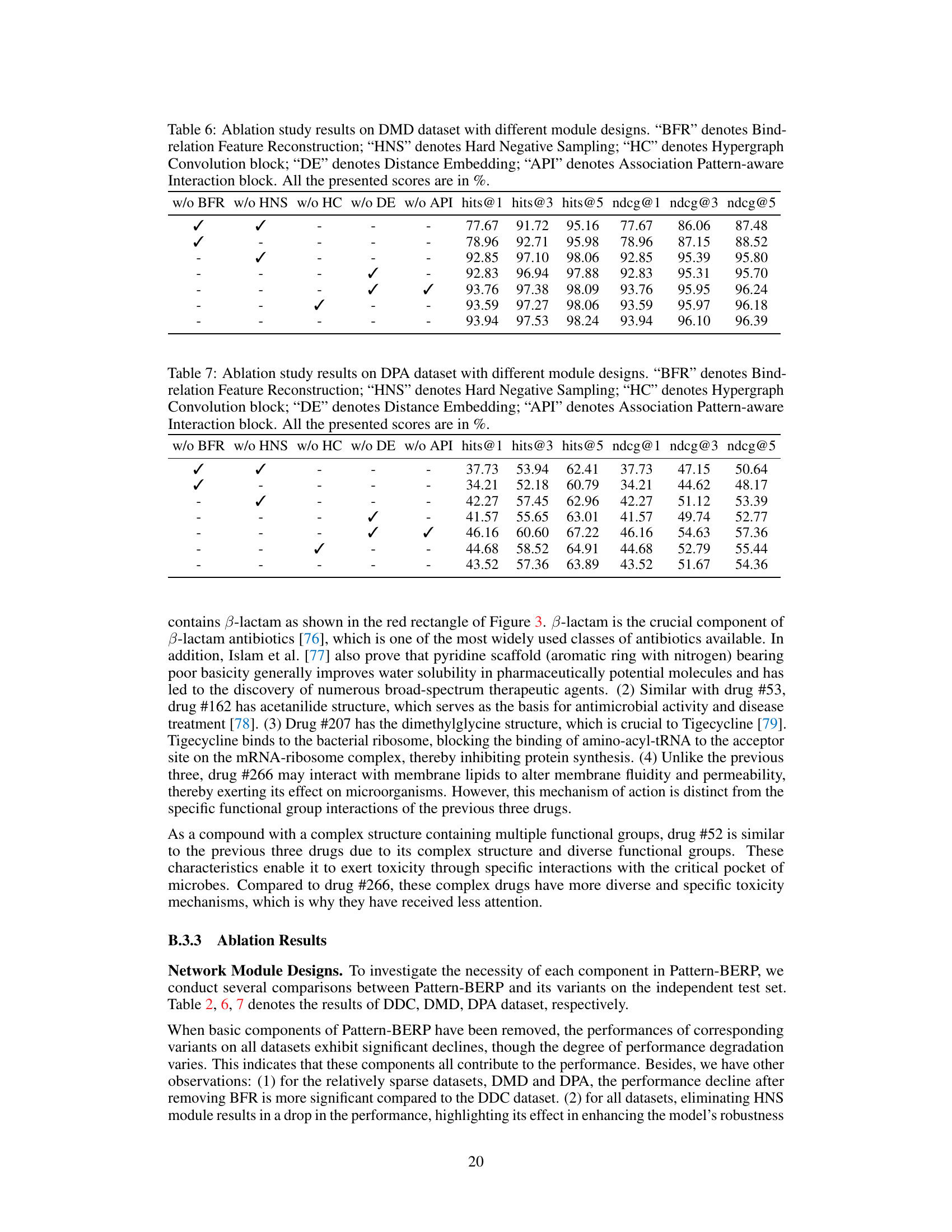
An ablation study systematically evaluates the contribution of individual components within a machine learning model. By removing or deactivating each part, researchers assess its impact on overall performance. This helps determine which parts are essential and which are redundant or even detrimental. In the context of a biological entity relationship prediction model, an ablation study might investigate the effects of removing modules responsible for specific tasks such as association pattern extraction, bind-relation enhancement, or hypergraph encoding. The results reveal the relative importance of each component and guide future improvements, highlighting features crucial for prediction accuracy and efficiency. A well-designed ablation study enhances the model’s interpretability by demonstrating the function of each part. Furthermore, by comparing the performance of the complete model to those with various components removed, it is possible to quantify the effect of each component and prioritize efforts towards enhancing the most impactful sections of the model.
Future Work#
The ‘Future Work’ section of this research paper presents exciting opportunities to expand upon the achievements detailed within. Extending the model to handle variable-length association patterns is crucial, as biological relationships are rarely confined to simple, fixed-length paths. This would involve exploring advanced graph traversal techniques or sequence modeling methods. Investigating the model’s performance in other domains beyond biology (e.g., knowledge graph completion, social network analysis) is another avenue to explore. This would involve adapting the model’s architecture and training strategies to the unique characteristics of each new domain. Addressing the computational complexity of the current model is necessary for scaling to even larger datasets. Techniques like graph sampling and more efficient attention mechanisms should be investigated. Finally, a deeper exploration of the biological interpretability of the association patterns discovered should be undertaken. This could include collaborations with biologists to validate findings and refine the biological interpretations provided.
More visual insights#
More on figures
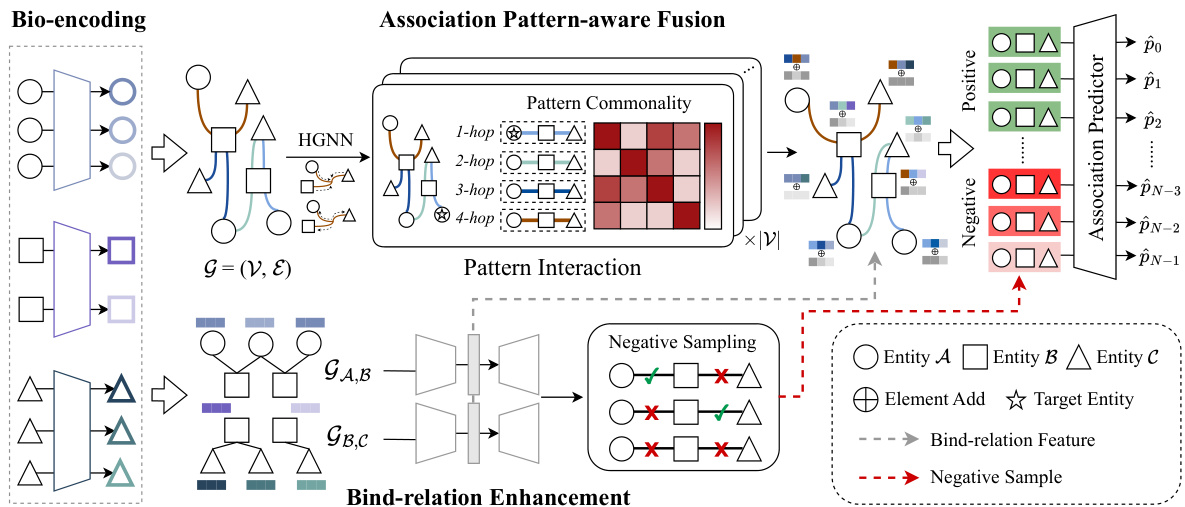
The figure illustrates the Pattern-BERP framework, which consists of four main modules: bio-encoding, association pattern-aware fusion, bind-relation enhancement, and association predictor. The bio-encoding module initializes entity attributes using different encoders based on entity type. A hypergraph and two bipartite graphs representing relationships are then constructed and fed into the association pattern-aware fusion module. This module incorporates association patterns to enhance entity representations. The bind-relation enhancement module reconstructs missing bind-relation features to generate hard negative samples. Finally, the association predictor module uses the combined entity features to predict the association.
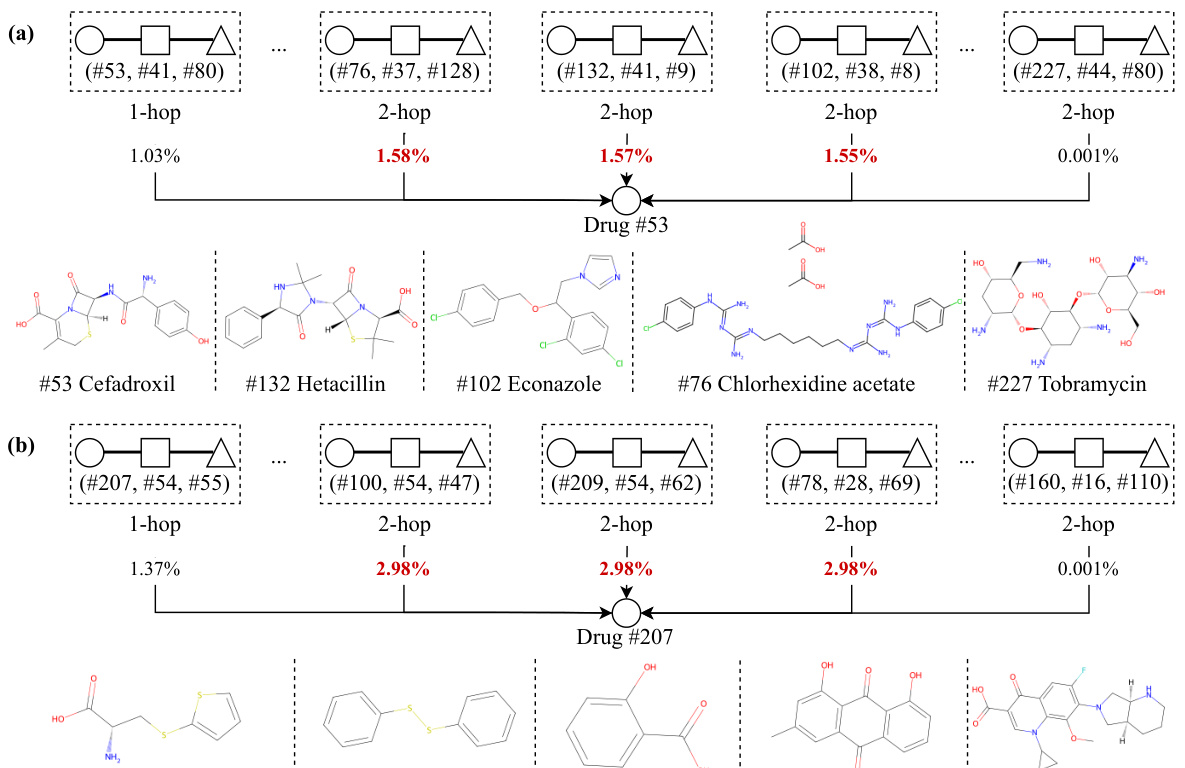
This figure visualizes the interpretability of the Pattern-BERP model by showing the association patterns for two drugs (Cefadroxil and S-(2-Thienyl)-L-cysteine) in the DMD dataset. The size of the node indicates the contribution of that pattern. Larger nodes indicate more significant contributions and similar biological pathways, while smaller nodes indicate less relevance.

This figure presents the overall architecture of the Pattern-BERP model. It starts by encoding the input entities (drugs, microbes, diseases) using different bio-encoders tailored to each entity type. Then, it builds a hypergraph and two bipartite graphs to represent the relationships between the entities. The hypergraph is processed by an Association Pattern-aware Fusion module which integrates association pattern information to enhance entity representation. A Bind-relation Enhancement module, based on the bipartite graphs, reconstructs missing information and generates hard negative samples for improved model training. Finally, an Association Predictor uses the integrated entity features to predict new entity relationships.

This figure illustrates the overall framework of the proposed Pattern-BERP model. It shows how entity attributes are encoded, how a hypergraph and two bipartite graphs are constructed from the data, and how these graphs are used in the Association Pattern-aware Fusion module and Bind-relation Enhancement module to generate an integrated entity feature for final association prediction. The process involves identifying association patterns, integrating them into the entity representation, and generating hard negative samples to improve the model’s performance.

This figure illustrates the overall architecture of the Pattern-BERP model. It shows how entity attributes are initialized, how the hypergraph and bipartite graphs are constructed, and how the association pattern-aware fusion and bind-relation enhancement modules are used to generate entity representations for association prediction.
This figure illustrates the overall framework of the Pattern-BERP model. It starts with bio-encoding of entities, then uses association pattern-aware fusion and bind-relation enhancement to generate final entity representation for association prediction. The figure shows how different components such as hypergraph, bipartite graphs and pattern interaction work together.
More on tables
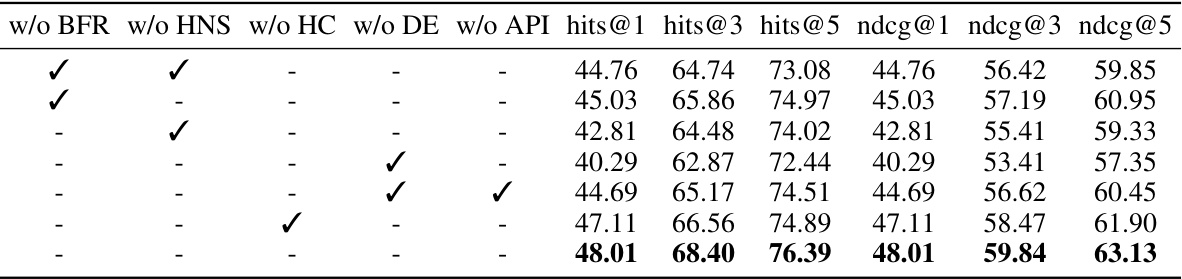
This table presents the ablation study results on the DDC dataset. It shows the impact of removing each component (Bind-relation Feature Reconstruction, Hard Negative Sampling, Hypergraph Convolution, Distance Embedding, and Association Pattern-aware Interaction) on the model’s performance. The performance metrics used are hits@1, hits@3, hits@5, ndcg@1, ndcg@3, and ndcg@5, all represented as percentages. The best results for each metric are highlighted in bold, showing the contribution of each module to the overall performance.
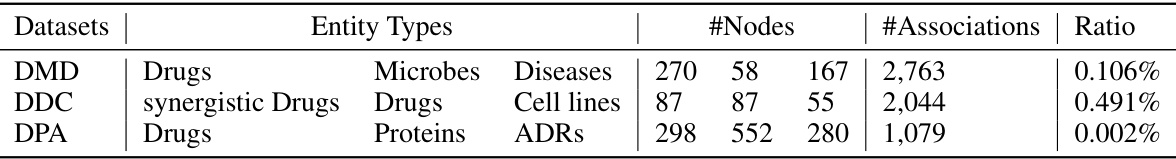
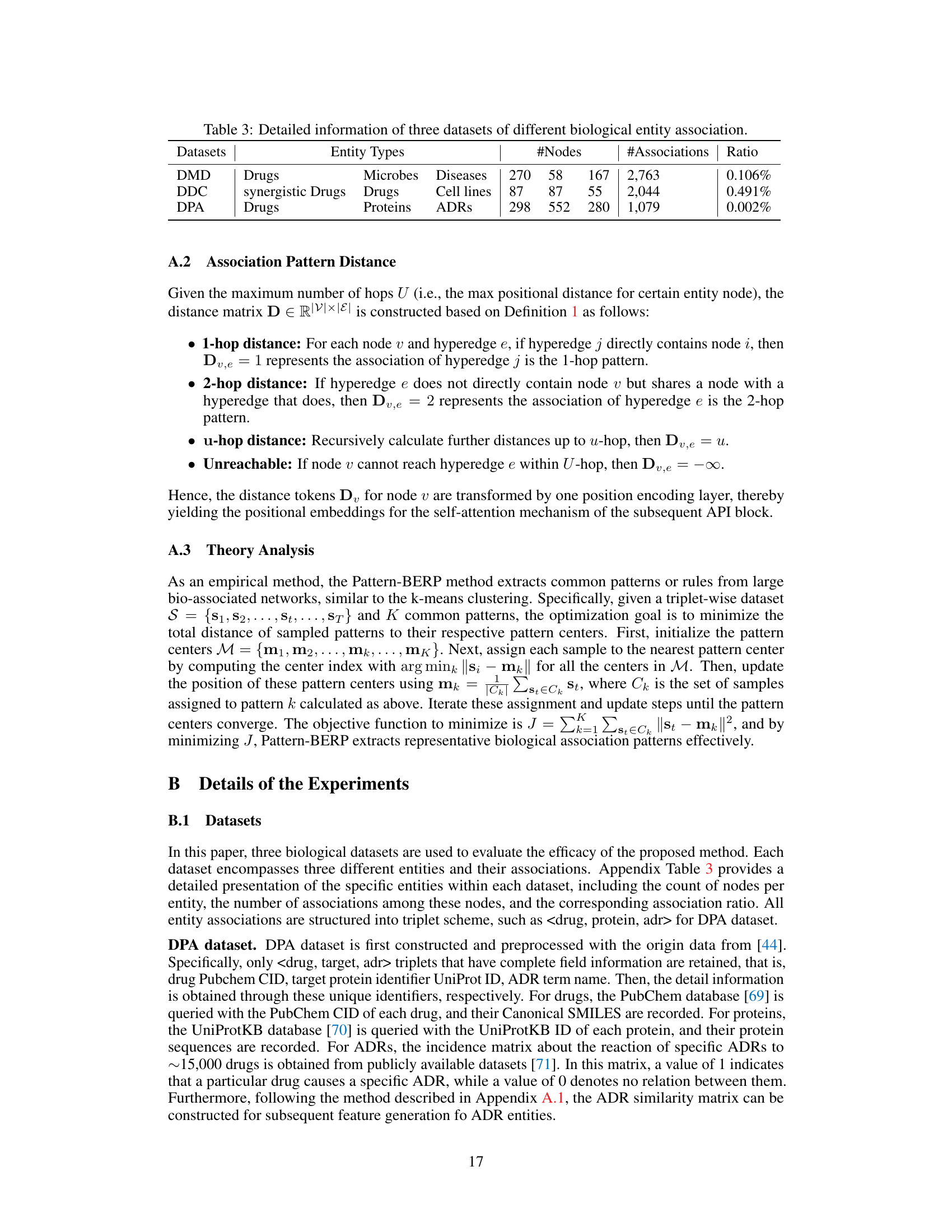
This table presents a detailed breakdown of three distinct biological entity association datasets used in the study: DMD (Drug-Microbe-Disease), DDC (synergistic Drug-Drug-Cell line), and DPA (Drug-target Protein-Adverse reaction). For each dataset, it lists the entity types involved, the number of nodes for each entity type, the total number of associations present, and the overall association ratio (the percentage of possible associations that are actually observed). This table provides crucial context for understanding the scale and characteristics of the data used in the experiments.

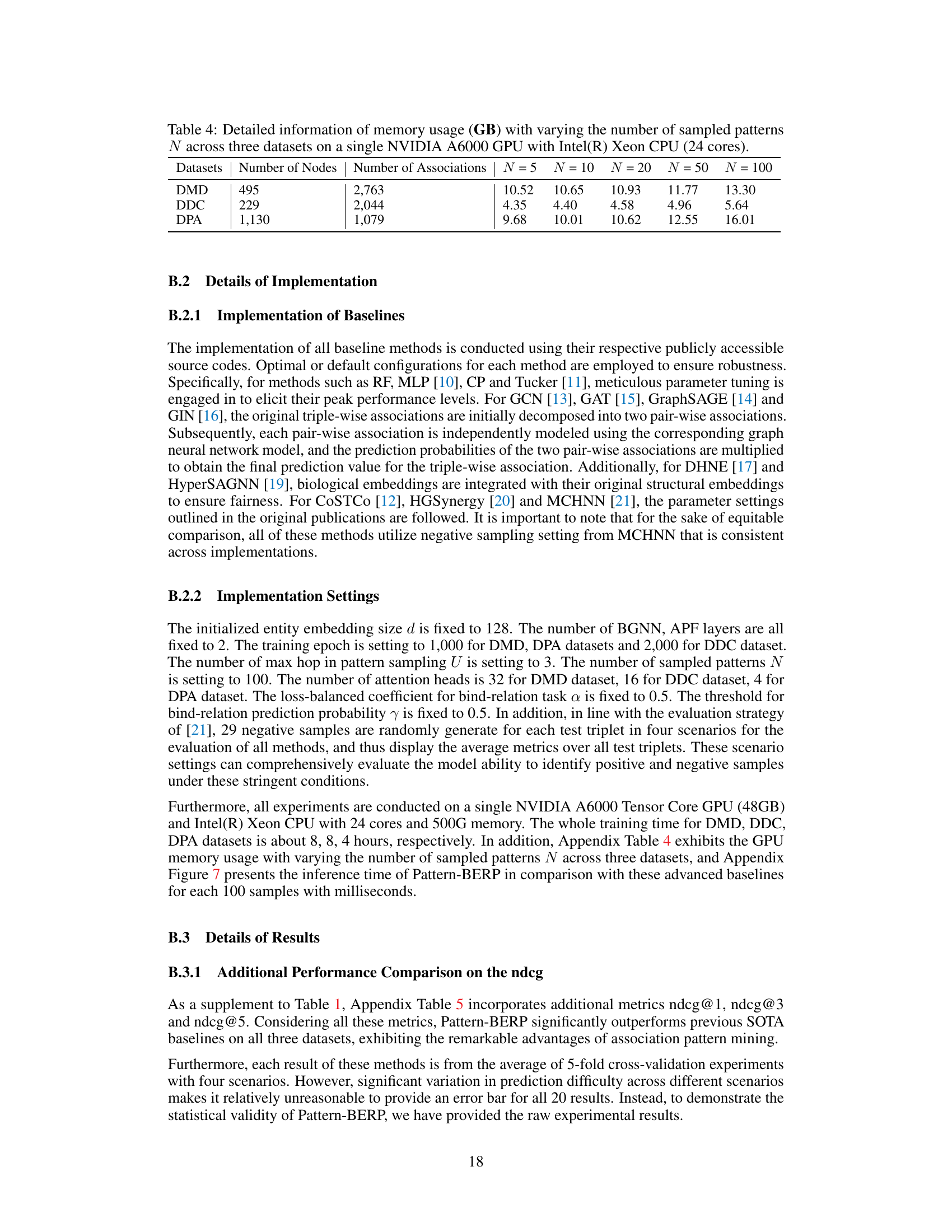
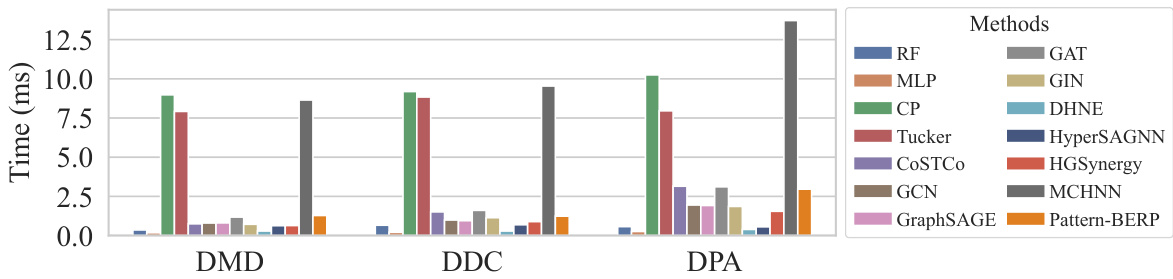
This table shows the GPU memory usage (in GB) for different numbers of sampled patterns (N = 5, 10, 20, 50, 100) across three biological datasets (DMD, DDC, DPA) using a single NVIDIA A6000 GPU and an Intel Xeon CPU with 24 cores. It demonstrates the scalability of the Pattern-BERP model in terms of memory consumption as the number of sampled patterns increases.
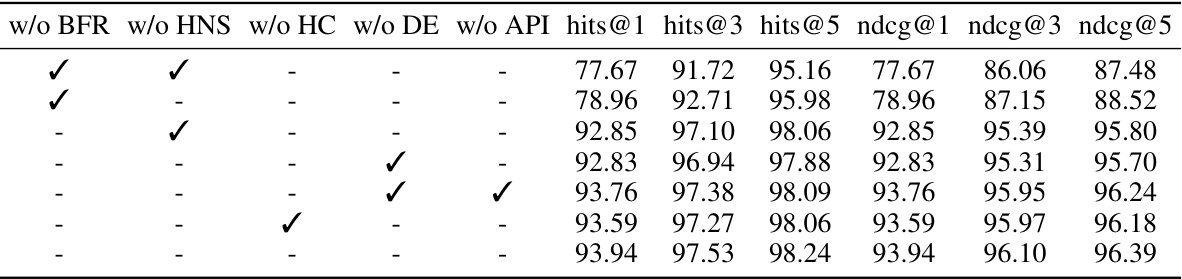
This ablation study evaluates the impact of each module (Bind-relation Feature Reconstruction, Hard Negative Sampling, Hypergraph Convolution, Distance Embedding, and Association Pattern-aware Interaction) in the Pattern-BERP model on the DDC dataset. The table shows the performance (hits@1, hits@3, hits@5, ndcg@1, ndcg@3, ndcg@5) when one module is removed. This helps understand the contribution of each component to the model’s overall performance.
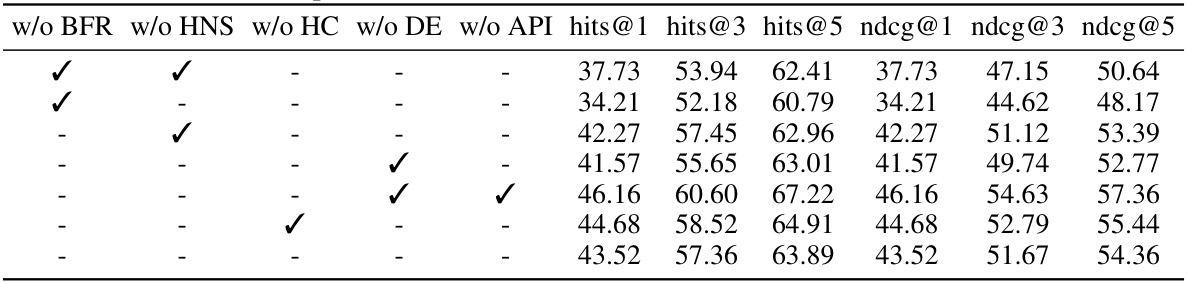
This ablation study shows the impact of removing different components (Bind-relation Feature Reconstruction, Hard Negative Sampling, Hypergraph Convolution, Distance Embedding, and Association Pattern-aware Interaction) of the Pattern-BERP model on the DPA dataset. The results, presented as percentages, reveal the contribution of each module to the overall performance, measured by hits@1, hits@3, hits@5, ndcg@1, ndcg@3, and ndcg@5.
Full paper#