↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Predicting how molecules affect cells is a major challenge in drug discovery. Traditional methods are slow and expensive, relying on extensive laboratory experiments. This paper tackles this challenge by using a new machine learning model that connects the molecular structure of a drug candidate to its impact on cell morphology, which is captured via microscopy images. The main issue is that experimental data is limited, has batch effects, and includes many inactive molecules. These difficulties hinder the development of an effective machine-learning model.
The researchers developed a new model called “MolPhenix” to overcome these issues. It uses a pre-trained model to understand the cell images, a new loss function to handle inactive molecules, and incorporates the concentration of the drug candidate into the model. This strategy greatly improves the accuracy of identifying active molecules that impact cells, leading to an 8.1x improvement over previous leading methods. This advancement is promising for faster and more efficient drug screening.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in drug discovery and computational biology. It presents a novel approach to efficiently identify molecules impacting cell function, which can significantly accelerate drug development and reduce costs. The methodology is broadly applicable and opens new avenues for multi-modal learning research.
Visual Insights#

This figure illustrates the architecture of the MolPhenix model, highlighting the three key guidelines employed to improve contrastive phenomolecular retrieval. Guideline 1 uses uni-modal pretrained models for phenomics (MAE) and molecular data (MPNN) to leverage knowledge learned from large datasets and mitigate data scarcity issues. Guideline 2 addresses the challenge of inactive molecules by incorporating a soft-weighted sigmoid locked loss (S2L) that weighs similarities between samples, undersampling inactive pairs. Guideline 3 incorporates molecular concentration information explicitly and implicitly using S2L, enhancing the model’s ability to generalize across different concentrations.

This table compares the performance of CLOOME and MolPhenix models in a zero-shot setting. The top part of the table shows results with a matched number of seen samples, highlighting that MolPhenix achieves an 8.1x improvement over CLOOME for identifying active unseen molecules. The bottom section displays results when using the best hyperparameters obtained after extensive training, demonstrating further performance gains and emphasizing the key role of the pre-trained uni-modal phenomics model in MolPhenix’s success.
In-depth insights#
Contrastive Learning#
Contrastive learning, a self-supervised learning technique, is pivotal in the paper’s methodology for aligning molecular structures and cellular phenomes. The core idea is to learn a joint embedding space where similar samples (a molecule and its corresponding cellular image) are close together, while dissimilar samples are far apart. This is achieved by training a model to distinguish between similar and dissimilar pairs, thereby capturing complex relationships between molecular structure and cellular response. The paper highlights the effectiveness of contrastive learning in enabling zero-shot molecular identification from microscopy images, showcasing its potential for high-throughput drug discovery. Challenges associated with contrastive learning, like imbalanced datasets and batch effects, are also addressed through innovative loss functions and data augmentation strategies. The success of contrastive learning in this application suggests its broader utility for multi-modal learning tasks in biology and beyond. The paper’s focus on contrastive learning emphasizes its power in building effective models even with relatively scarce paired data, a crucial consideration in many biological domains where data acquisition can be costly and time-consuming.
Multimodal Phenomics#
Multimodal phenomics represents a significant advancement in the field of drug discovery and biological research. By integrating multiple data modalities, such as microscopy images, genomic data, and molecular structures, it enables a more holistic and comprehensive understanding of cellular responses to molecular perturbations. Contrastive learning is a powerful technique used to align these diverse data types, creating a shared latent space. This shared space allows researchers to make predictions about a molecule’s impact on cellular function (phenotype) based on its structure or predict the structure based on the effects it has on the cell. The development of novel loss functions, such as the soft-weighted sigmoid loss (S2L), has addressed the challenges of imbalanced data and inactive molecules significantly improving the accuracy of predictions. MolPhenix, as an example, demonstrates the strength of the multimodal phenomics approach through high accuracy in zero-shot molecular retrieval. This methodology has the potential to drastically improve drug discovery by reducing time and cost of experiments.
MolPhenix Model#
The MolPhenix model, as described in the research paper, presents a novel approach to contrastive pheno-molecular retrieval. It leverages a pretrained uni-modal phenomics model (Phenom1) to generate robust representations of cellular morphology, mitigating the impact of batch effects and reducing the need for large datasets. A key innovation is the introduction of a novel soft-weighted sigmoid locked loss (S2L), which addresses the challenges posed by inactive molecules and improves the model’s ability to learn inter-sample similarities. Furthermore, MolPhenix incorporates methods to explicitly and implicitly encode molecular concentration, enhancing the model’s capacity to generalize across different dosage levels and leading to a significant improvement in zero-shot molecular retrieval accuracy. The architecture elegantly combines these components, demonstrating a significant leap in the field of virtual phenomics screening and drug discovery. The model’s ability to perform zero-shot retrieval, across varying concentrations and molecular scaffolds suggests significant potential for future applications.
Zero-Shot Retrieval#
Zero-shot retrieval, a crucial aspect of the research, focuses on the model’s ability to identify molecular structures based solely on phenomic experimental data without prior training on those specific molecules. This capability is demonstrated as a significant advancement, enabling the model to generalize to unseen molecules. The success of zero-shot retrieval highlights the model’s learned understanding of the relationship between molecular structures and their impact on cellular morphology. Improved multi-modal learning techniques like contrastive learning and a novel inter-sample similarity loss function significantly enhance the model’s capacity for zero-shot retrieval. A key challenge addressed is the handling of inactive molecules, which are perturbations not affecting cell morphology and thus misaligned with experimental data; techniques such as soft-weighted sigmoid locked loss (S2L) are developed to address this issue. The remarkable retrieval accuracy achieved (77.33% in top 1%) in the zero-shot setting underscores the robustness and effectiveness of the model and opens opportunities for virtual phenomics drug discovery applications.
Future Directions#
The paper’s ‘Future Directions’ section would ideally explore several key avenues. Expanding to other modalities beyond microscopy images and molecular structures (e.g., incorporating genomic data, transcriptomics, or chemical multi-compound interventions) would significantly enrich the model’s predictive power and biological relevance. Addressing the challenges of batch effects more robustly through advanced normalization techniques or domain adaptation methods should be a priority. Furthermore, a deeper exploration of implicit and explicit concentration encoding strategies is needed to determine optimal representations. The study should investigate the model’s generalizability across diverse cell types and experimental conditions, and perhaps assess its capabilities through wet-lab validation, comparing in-silico predictions against real-world experimental data. Finally, thorough investigation into the model’s limitations regarding specific biological mechanisms and the potential for bias due to dataset composition would build trust and improve interpretability. Addressing these points will significantly advance the field of contrastive pheno-molecular retrieval.
More visual insights#
More on figures
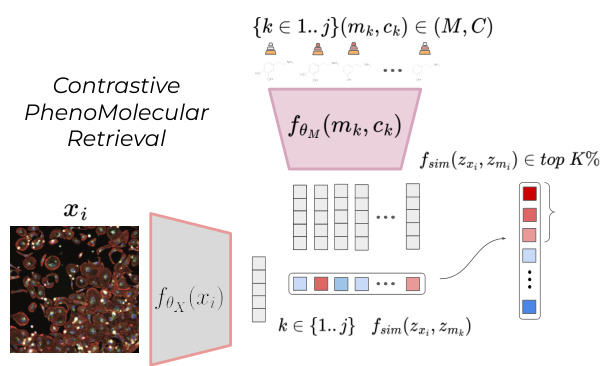
This figure illustrates the core challenge addressed in the paper: contrastive phenomolecular retrieval. Given a microscopy image (xi) showing a cell’s morphology after treatment with a molecule, the task is to identify which molecule (from a set of candidates {mk, ck}) caused the observed morphological change. The model learns a joint embedding space for both image and molecular data. The similarity between the image embedding (zxi) and each molecule embedding (zm) is calculated using a similarity metric (fsim). The correct molecule should ideally rank highly (within the top K%) in terms of similarity.

This figure illustrates the contrastive phenomolecular retrieval challenge. Given a microscopy image (xᵢ) of cells exhibiting a morphological change, the goal is to identify the molecule (mᵢ) and its concentration (cᵢ) that caused the change from a set of possible molecules and concentrations. The process involves embedding both the image and each molecule-concentration pair into a shared latent space (Rd). A similarity metric (fsim) is used to compare the image embedding with each molecule-concentration pair embedding and rank the pairs based on similarity. A successful retrieval occurs if the correct molecule and concentration are ranked within the top K%.
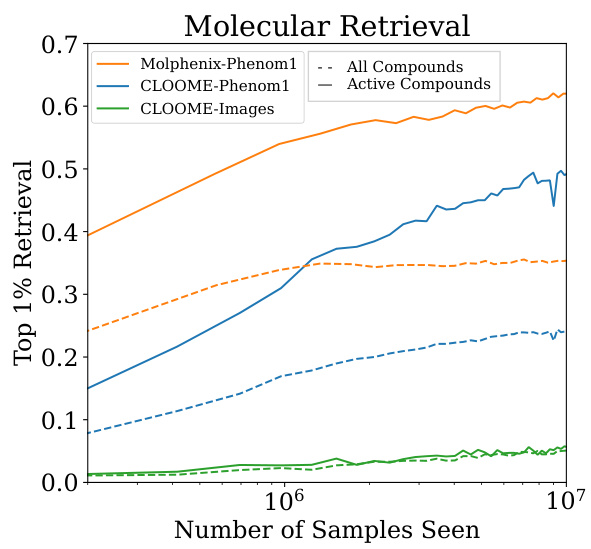
This figure compares the performance of three different models on a molecular retrieval task: MolPhenix trained with Phenom1, CLOOME trained with Phenom1, and CLOOME trained with images. The x-axis shows the number of samples seen during training, plotted on a logarithmic scale to better visualize the progression. The y-axis represents the top 1% retrieval accuracy. Two lines are shown for each model, one for all compounds and one for only active compounds, revealing the performance differences between these two groups. The figure demonstrates that MolPhenix trained with the pre-trained Phenom1 model significantly outperforms the other two models, highlighting the effectiveness of utilizing a pre-trained phenomics encoder. The gap between the performance on all compounds and active compounds is also notable for each model.

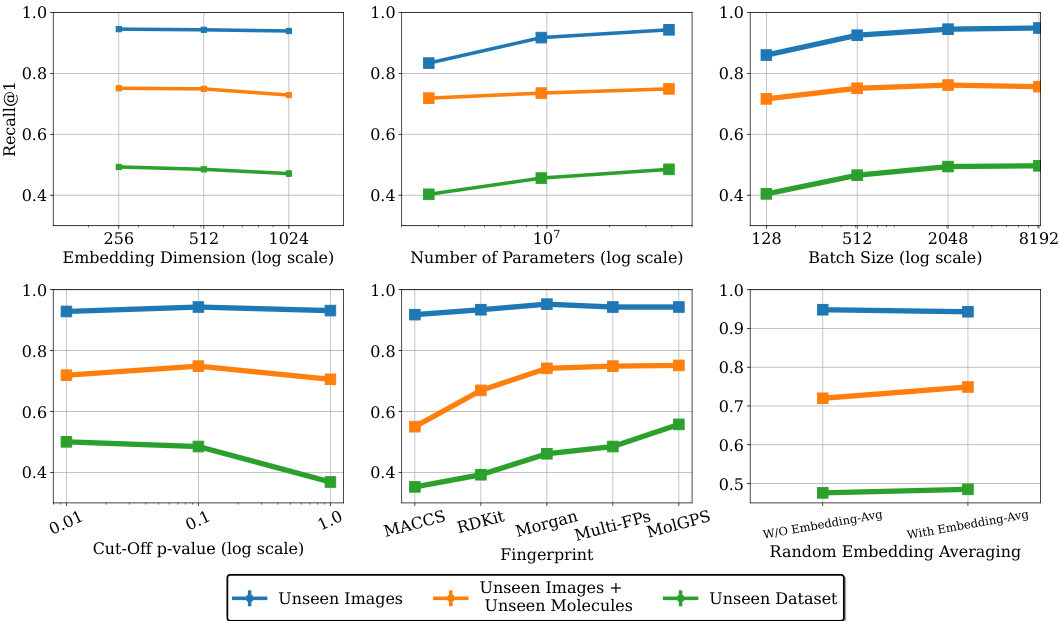
This figure shows the ablation study on MolPhenix, examining the effects of different hyperparameters and design choices on the model’s performance. It demonstrates the positive impact of using compact embeddings from pre-trained models, larger numbers of parameters, larger batch sizes, lower cutoff p-values, using pretrained MolGPS fingerprints, and incorporating random batch averaging.
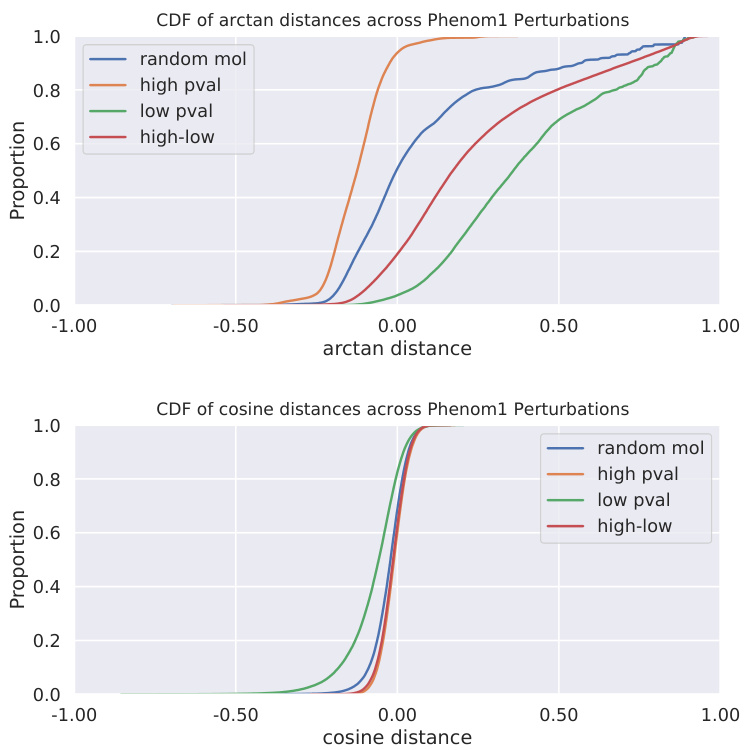
This figure shows the cumulative distribution functions (CDFs) of two distance metrics (cosine similarity and arctangent of L2 distance) calculated between Phenom1 embeddings. Four groups of molecule pairs are compared: those with random molecule selection, those where both molecules are active (low p-value), those where both are inactive (high p-value), and those with one active and one inactive molecule (high-low). The comparison highlights the difference in distribution separability between cosine similarity and the arctangent of L2 distance, particularly for distinguishing between active and inactive molecules. The arctangent of L2 distance shows a better separation between these groups, suggesting that it’s a more effective metric for this task.

This figure shows the results of dimensionality reduction on the chemical embeddings from the unseen RXRX3 dataset using UMAP. The points are colored according to their activity (p-values from Phenom1). The top panel shows all embeddings together, while the bottom panels show separate UMAPs for different concentrations.
This figure illustrates the core challenge of contrastive phenomolecular retrieval. A single phenomic image (x₁) is presented along with a set of molecules (mk) and their associated concentrations (ck). The goal is for a model to learn a joint latent space where the image and molecule/concentration pairs are meaningfully related. The model should be able to identify (within the top K%) the correct molecule/concentration pair (mk, ck) that corresponds to the observed effects in image x₁. This zero-shot retrieval task requires the model to effectively capture cross-modal relationships between phenomic images and molecular representations, highlighting the core challenge of the paper.
This figure displays the results of dimensionality reduction using UMAP on chemical embeddings from the unseen RXRX3 dataset. The points are colored by their activity (p-values), showing distinct clusters for different activity levels. The top panel shows the overall distribution, while the bottom panels display the distributions for specific concentrations (0.25µM, 1.0µM, 2.5µM, 10.0µM). This visualization helps to understand how well the model separates active and inactive molecules based on their chemical features and concentration.

This figure illustrates the architecture of MolPhenix, a contrastive phenomolecular retrieval model. The model incorporates three key guidelines to improve performance: 1) using pre-trained uni-modal models for phenomics (MAE) and molecular (MPNN) data; 2) employing a novel soft-weighted sigmoid locked loss (S2L) that addresses the effects of inactive molecules and uses inter-sample similarities; and 3) explicitly encoding molecular concentration. The figure shows how these components work together to generate molecular and phenomic embeddings that are compared to achieve contrastive learning and accurate retrieval.
This figure illustrates the architecture of the MolPhenix model, highlighting three key guidelines for improved performance: using pretrained unimodal models for phenomics and molecular data, incorporating a novel inter-sample similarity-aware loss function (S2L) to handle inactive molecules, and encoding molecular concentration explicitly. The diagram shows the flow of data through the model, from input phenomics experiments and molecular structures, to the final similarity logits used for retrieval.
This figure illustrates the architecture of the MolPhenix model, a contrastive phenomolecular retrieval framework. It highlights three key guidelines to improve retrieval performance: (1) utilizing uni-modal pretrained models for phenomics (MAE) and molecular (MPNN) data; (2) employing a novel soft-weighted sigmoid locked loss (S2L) to address inactive molecules and inter-sample similarities; and (3) encoding molecular concentration explicitly and implicitly within the S2L loss. The diagram shows the flow of data through the various components of the model, emphasizing the integration of these guidelines.
More on tables
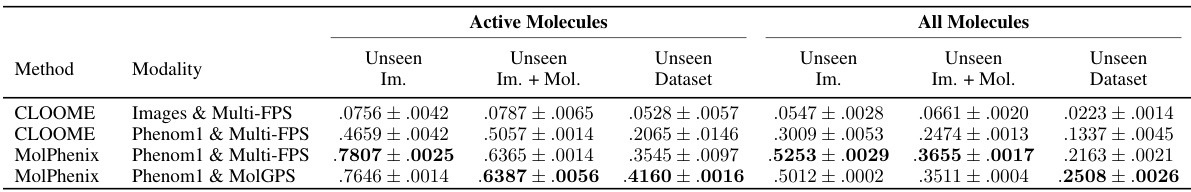
This table compares the performance of CLOOME and MolPhenix models on the task of zero-shot molecular retrieval. The results are broken down into different categories: active molecules (molecules known to have a biological effect), all molecules, and for unseen images (images of cells treated with molecules not seen during training), unseen images + unseen molecules (zero-shot molecular retrieval), and unseen dataset (zero-shot molecular retrieval on an independent test dataset). The table shows significant improvements in performance achieved by MolPhenix, particularly when using both pre-trained phenomics and molecular models and the novel S2L loss function. The bottom section shows results with optimal hyperparameters, revealing an 8.1x improvement over the state-of-the-art. Note that the effectiveness of MolPhenix relies on using a pre-trained uni-modal phenomics model.
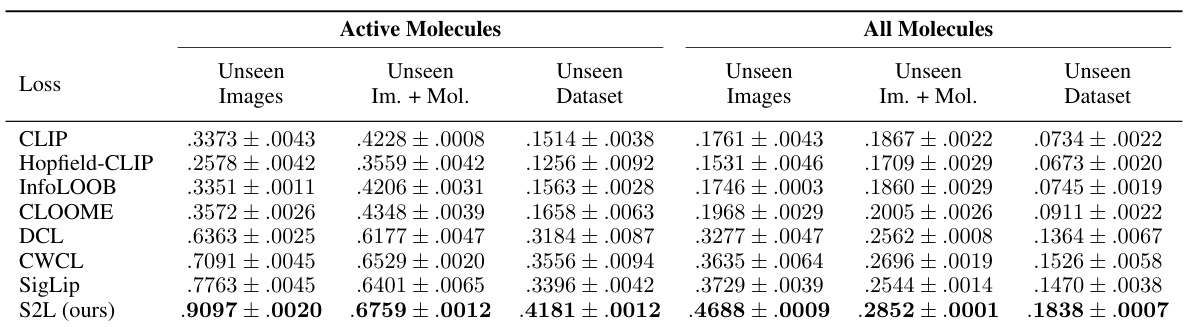
This table compares the performance of CLOOME and MolPhenix models on several tasks, highlighting the significant improvement achieved by MolPhenix, particularly for active unseen molecules (8.1x better than CLOOME). The impact of using pre-trained uni-modal models (Phenom1 and MolGPS) is also shown. The table also presents state-of-the-art (SOTA) results achieved with additional training.

This table compares the performance of CLOOME and MolPhenix models on three different datasets. The top part shows results for the matched number of seen samples, highlighting MolPhenix’s 8.1x improvement in top-1% retrieval accuracy over CLOOME for active unseen molecules. The bottom section presents state-of-the-art (SOTA) results achieved with more training steps and optimal hyperparameters, emphasizing MolPhenix’s reliance on a pre-trained uni-modal phenomics model for its improved performance.

This table presents the top-1% recall accuracy results achieved by different methods on unseen images and unseen molecules, along with the overall performance on unseen datasets. The results highlight the improvement in accuracy obtained by incorporating the proposed MolPhenix guidelines, namely utilizing a pre-trained phenomics model (Phenom1) and averaging embeddings. The experiment omits explicit concentration encoding.

This table compares the performance of CLOOME and MolPhenix models on active and all molecules, with different modalities (images, Phenom1, and MolGPS), showing a significant improvement in MolPhenix’s performance, especially for active unseen molecules. It highlights the impact of using pre-trained uni-modal encoders, undersampling inactive molecules, and encoding molecular concentrations.
This table compares the performance of CLOOME and MolPhenix models on various tasks, demonstrating MolPhenix’s significant improvements, especially regarding active molecule retrieval. The top section shows results with a matched number of seen samples, highlighting MolPhenix’s superior performance. The bottom section shows state-of-the-art results achieved by training with a higher number of steps. It emphasizes the importance of MolPhenix’s key components—S2L loss and embedding averaging—which rely on a pre-trained uni-modal phenomics model.

This table compares the performance of CLOOME and MolPhenix models on active and all molecules under various conditions. It highlights the significant performance gain achieved by MolPhenix (8.1x improvement over CLOOME) when using a pre-trained Phenom1 model and MolGPS embeddings. The table also shows results for both a matched number of seen samples and state-of-the-art (SOTA) results with more training steps, emphasizing the impact of pre-trained models and the proposed MolPhenix techniques.

This table presents a comparison of the performance of CLOOME and MolPhenix models on unseen molecules. The top section shows the results for a matched number of seen samples, highlighting the 8.1x improvement achieved by MolPhenix over CLOOME for active molecules. The bottom section displays the state-of-the-art results obtained by training with a larger number of steps and optimal hyperparameters, demonstrating MolPhenix’s consistent superior performance. The table also notes the reliance of MolPhenix’s key components (S2L loss and embedding averaging) on a pre-trained uni-modal phenomics model.
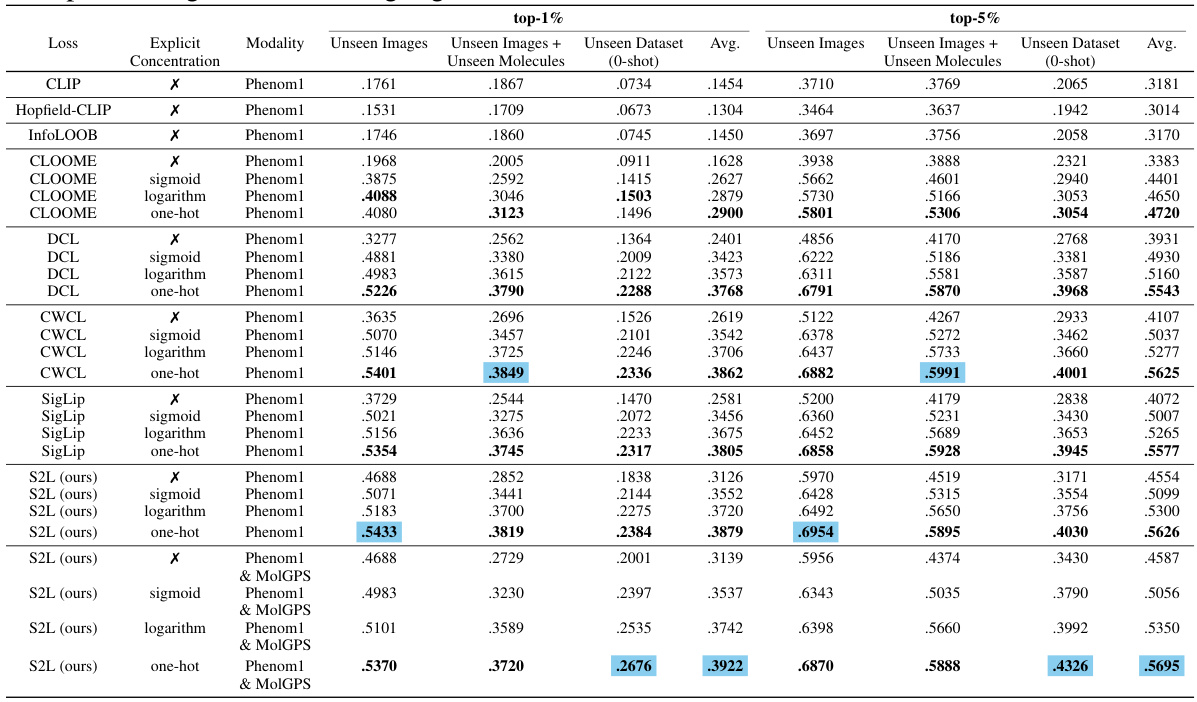
This table presents a comparison of the performance of CLOOME and MolPhenix models on unseen data, specifically focusing on the top 1% and top 5% retrieval accuracy for both active and all molecules. The results highlight the significant performance improvement achieved by MolPhenix (8.1x improvement over CLOOME for active molecules) through the utilization of a pre-trained uni-modal phenomics model and other methodological improvements.
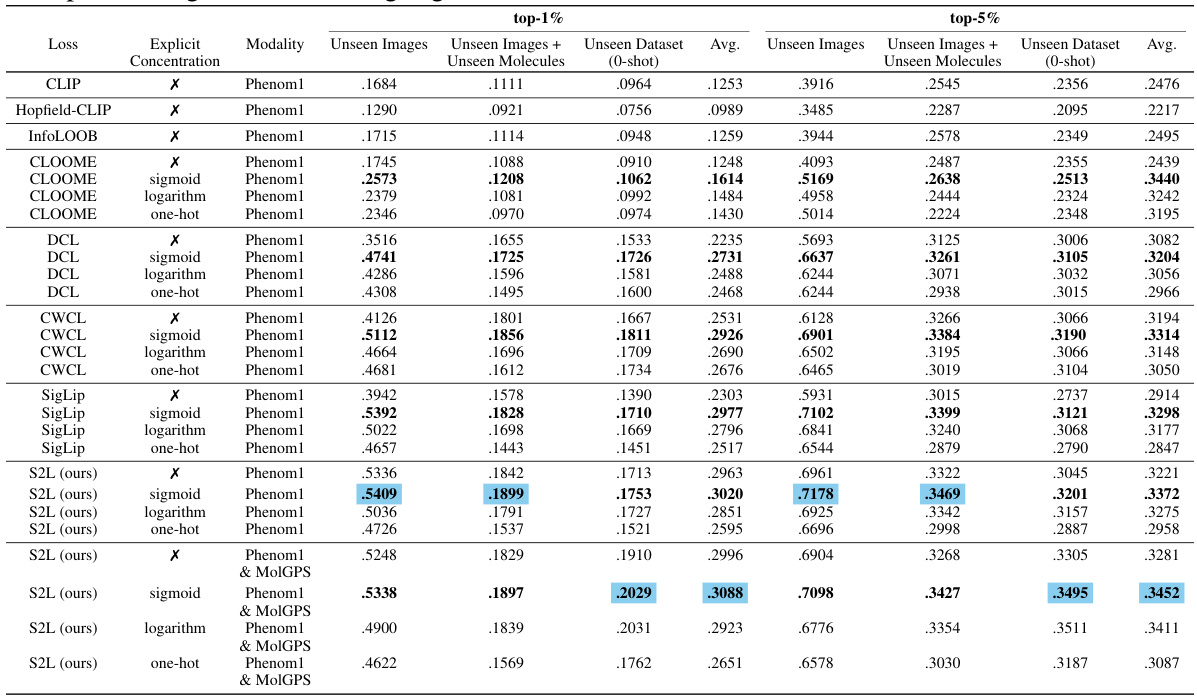
This table compares the performance of CLOOME and MolPhenix models on different tasks, using various modalities (images, Phenom1, and MolGPS). The top part shows the results for a matched number of seen samples, highlighting the 8.1x improvement of MolPhenix over CLOOME for active unseen molecules. The bottom part shows the state-of-the-art results, obtained by training with more steps and using optimal hyperparameters. The table emphasizes that MolPhenix’s performance gain relies on using a pre-trained uni-modal phenomics model (Phenom1) and incorporating techniques like S2L loss and embedding averaging.

This table compares the performance of CLOOME and MolPhenix models on various tasks, using different modalities (images, Phenom1, and MolGPS). The top section shows results with a matched number of seen samples, highlighting the significant improvement of MolPhenix (8.1x) over CLOOME for active, unseen molecules. The bottom section presents state-of-the-art (SOTA) results achieved with more training steps and optimal hyperparameters. It emphasizes that MolPhenix’s key improvements (S2L loss and embedding averaging) rely on using a pre-trained uni-modal phenomics model.
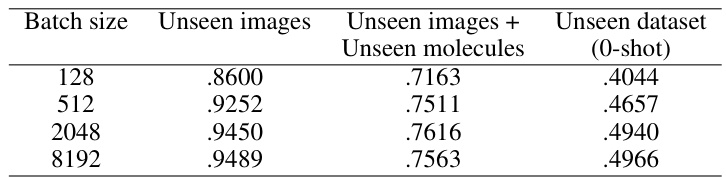
This table compares the performance of CLOOME and MolPhenix models on active and all molecules in different scenarios (unseen images, unseen images+molecules, unseen datasets). The top part shows the results for the matched number of seen samples, highlighting an 8.1x improvement of MolPhenix over CLOOME on active, unseen molecules. The bottom part displays SOTA results using optimized hyperparameters and a higher number of training steps, emphasizing the importance of MolPhenix’s components (S2L loss and embedding averaging) and the pretrained phenomics model.
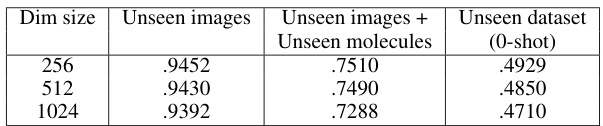
This table compares the performance of CLOOME and MolPhenix models on three different unseen datasets, showcasing MolPhenix’s significant improvement in zero-shot molecular retrieval. The top section shows results for a matched number of seen samples, highlighting MolPhenix’s 8.1x improvement over CLOOME for active unseen molecules. The bottom section presents state-of-the-art results achieved by training with a higher number of steps and optimal hyperparameters, further emphasizing MolPhenix’s superior performance.
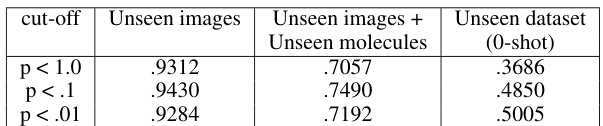
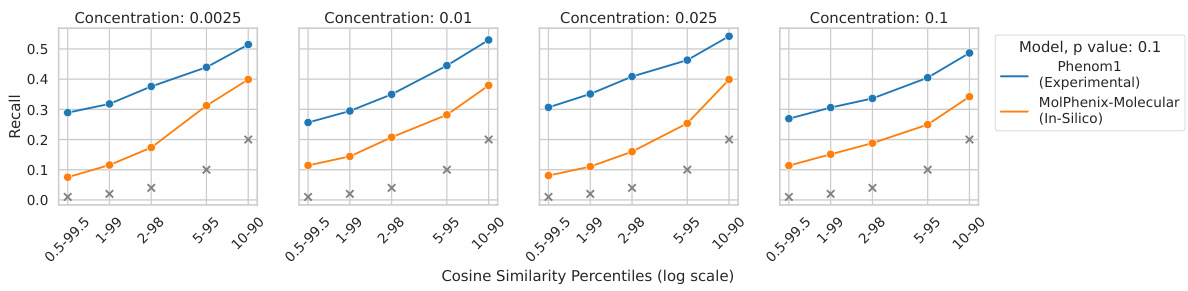
This ablation study investigates the impact of different p-value cutoffs on the retrieval performance of active molecules. The results show that using a p-value cutoff of less than 0.1 improves the retrieval performance of active molecules across different datasets.

This table compares the performance of CLOOME and MolPhenix models on different tasks, highlighting the impact of using pre-trained Phenom1 and MolGPS models. The top section shows results for a matched number of seen samples, demonstrating MolPhenix’s significant improvement over CLOOME, especially for active unseen molecules. The bottom section presents state-of-the-art (SOTA) results achieved with more training steps and optimal hyperparameters, further showcasing MolPhenix’s superior performance. The table emphasizes the importance of using a pre-trained uni-modal phenomics model and components like S2L and embedding averaging for MolPhenix’s success.

This table compares the performance of CLOOME and MolPhenix models on active and all molecules using different modalities (images, Phenom1, MolGPS). It highlights the significant improvement in zero-shot retrieval accuracy achieved by MolPhenix (8.1x improvement over CLOOME for active unseen molecules). It also shows that MolPhenix’s performance is highly dependent on the use of a pre-trained phenomics model and the proposed guidelines (uni-modal pretrained models, S2L loss, undersampling of inactive molecules, and encoding concentration).
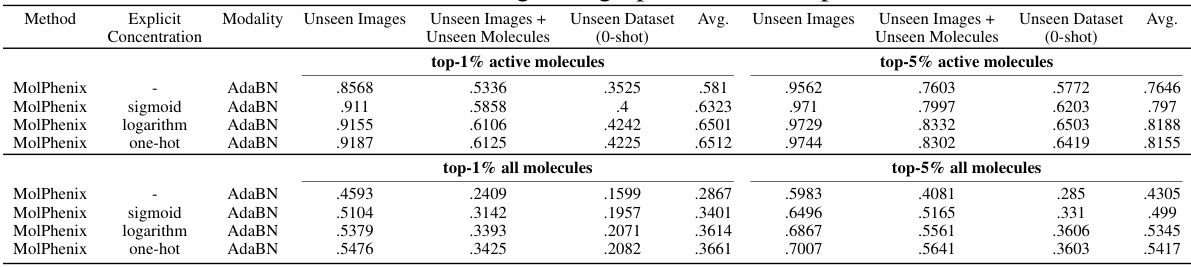
This table compares the performance of different models (CLOOME and MolPhenix) with various configurations (using different modalities like images, Phenom1, and MolGPS). It demonstrates a significant improvement in zero-shot molecular retrieval accuracy achieved by MolPhenix, especially for active unseen molecules, compared to the previous state-of-the-art (CLOOME). The table highlights the impact of using pre-trained uni-modal models and the novel techniques implemented in MolPhenix (like S2L loss and embedding averaging). The results are presented for both active and all molecules, and show that MolPhenix’s performance is consistent across various scenarios.
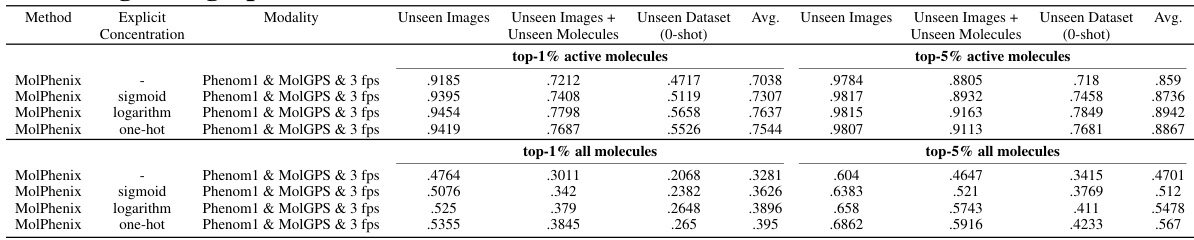
This table compares the performance of different models (CLOOME and MolPhenix) in a molecular retrieval task, considering different modalities for input (images and fingerprints) and the usage of pre-trained models for phenomic and molecular information. The top section shows results for a matched number of samples, highlighting the 8.1x improvement of MolPhenix over CLOOME for active unseen molecules. The bottom section presents state-of-the-art (SOTA) results achieved with a higher number of training steps and optimal hyperparameters. The table emphasizes that MolPhenix’s superior performance depends on its use of a pre-trained uni-modal phenomics model.

This table presents a comparison of the performance of CLOOME and MolPhenix models, with and without pre-trained models (Phenom1 and MolGPS), on various metrics (top-1% and top-5% recall accuracy for active and all molecules on unseen images, unseen images+molecules, and unseen datasets). The top section shows results with a matched number of seen samples, highlighting the significant improvement achieved by MolPhenix. The bottom section shows the state-of-the-art (SOTA) results obtained by training with more steps using optimal hyperparameters. The table emphasizes the contribution of MolPhenix’s key components, like S2L loss and embedding averaging, enabled by the pre-trained uni-modal phenomics model.
Full paper#