↗ arXiv ↗ Hugging Face ↗ Hugging Face ↗ Chat
TL;DR#
Predicting protein structures accurately requires high-quality Multiple Sequence Alignments (MSAs), which are often unavailable for many proteins. Existing methods for generating virtual MSAs have limitations in capturing intricate co-evolutionary patterns or require guidance from external models. This leads to compromised prediction accuracy, particularly for proteins with insufficient homologous sequences.
The paper introduces MSAGPT, a novel approach that uses generative pre-training to generate virtual MSAs for prompting protein structure predictions. MSAGPT incorporates a 2D evolutionary positional encoding scheme and a 1D MSA decoding framework, facilitating zero- or few-shot learning. Furthermore, it leverages feedback from AlphaFold2 to enhance the model’s capacity and reduce hallucinations. Extensive experiments demonstrate MSAGPT’s ability to generate faithful virtual MSAs, improving structure prediction accuracy significantly, especially in low-MSA scenarios. The transfer learning capabilities showcased highlight its potential for various protein-related tasks.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in protein structure prediction and related fields due to its novel approach to address the challenge of low-MSA data. MSAGPT’s innovative method of generating high-quality virtual MSAs significantly enhances prediction accuracy, opening exciting avenues for research on orphan proteins and advancing the field of structural biology. The transfer learning capabilities also broaden its impact across various protein-related tasks.
Visual Insights#
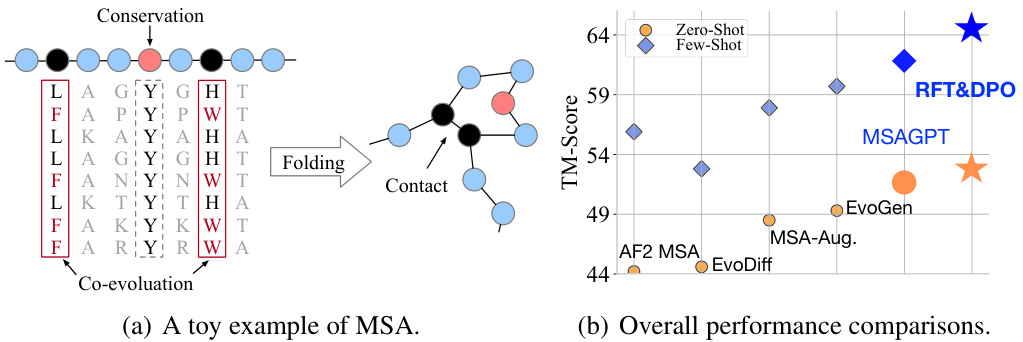
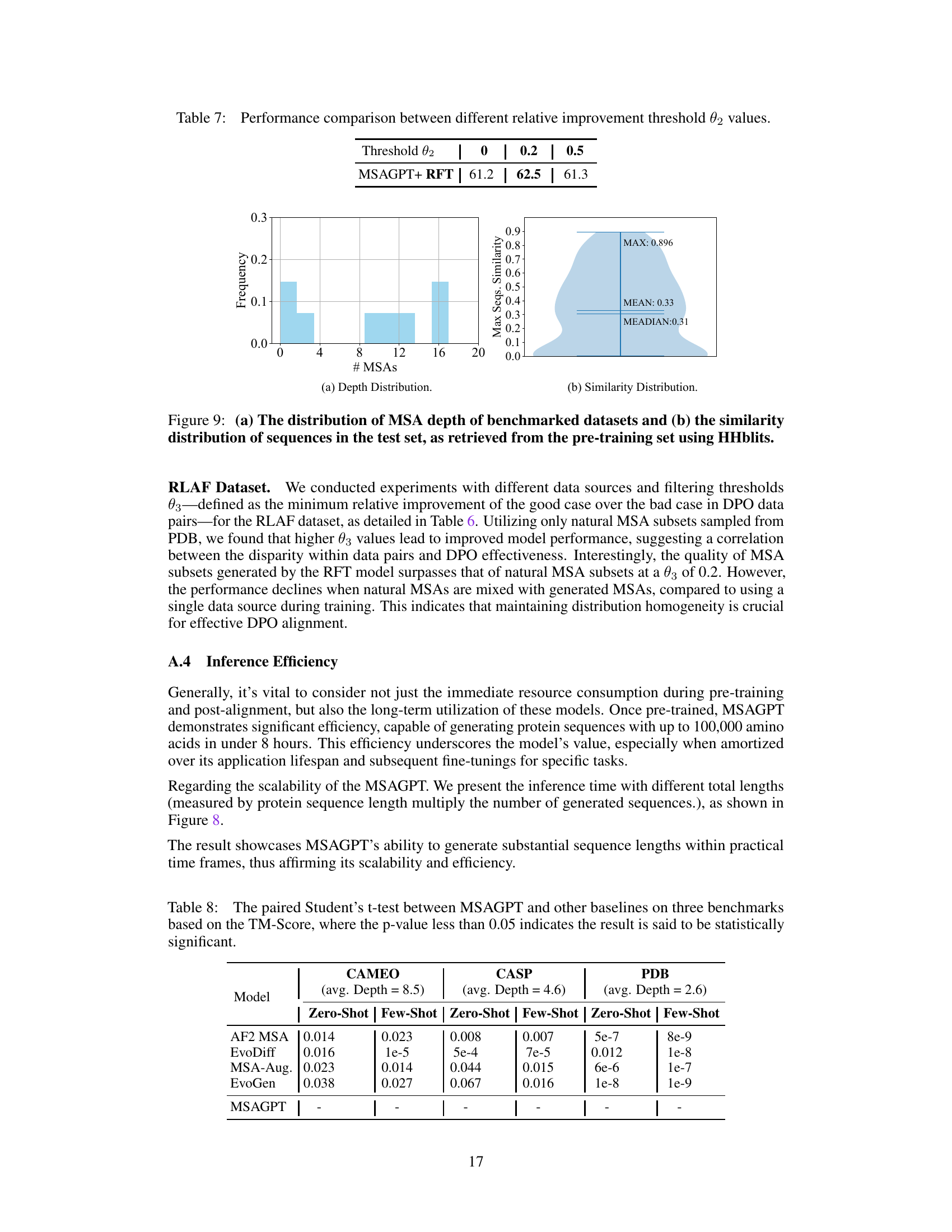
🔼 Figure 1(a) demonstrates a toy example of MSA, illustrating the co-evolutionary patterns within the MSA. This helps understand the relationships between amino acid sites that influence the folding structures. Figure 1(b) presents the overall performance comparison between MSAGPT and advanced baselines on three natural MSA-scarce benchmarks. It highlights MSAGPT’s superior performance in protein structure prediction tasks, especially when limited MSA is available.
read the caption
Figure 1: (a) The illustration of MSA and (b) performance comparisons between MSAGPT and advanced baselines on three natural MSA-scarce benchmark.
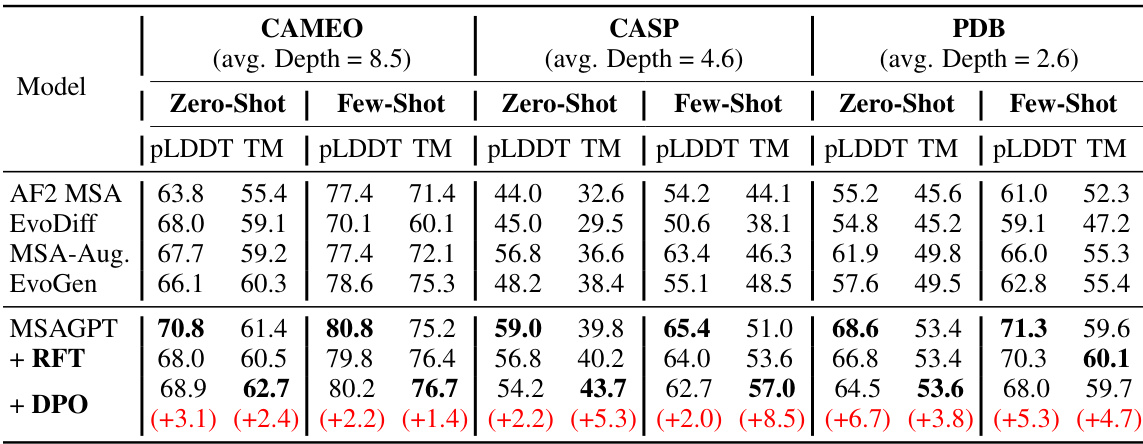
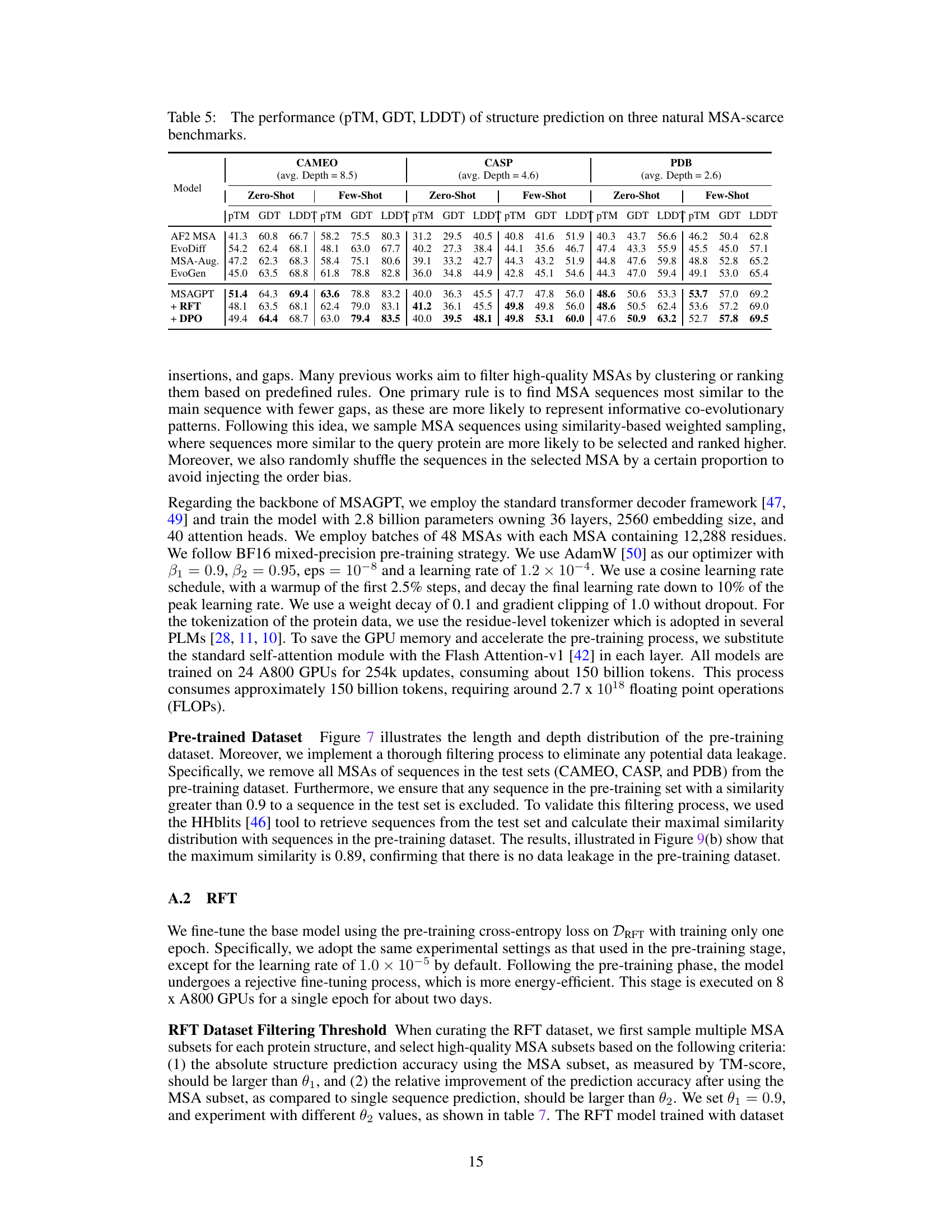
🔼 This table presents the performance comparison of different models on three benchmark datasets with limited MSA information. The models are evaluated using several metrics, including PLDDT and TM-score. The results show that MSAGPT, particularly after RFT and DPO, outperforms baseline methods.
read the caption
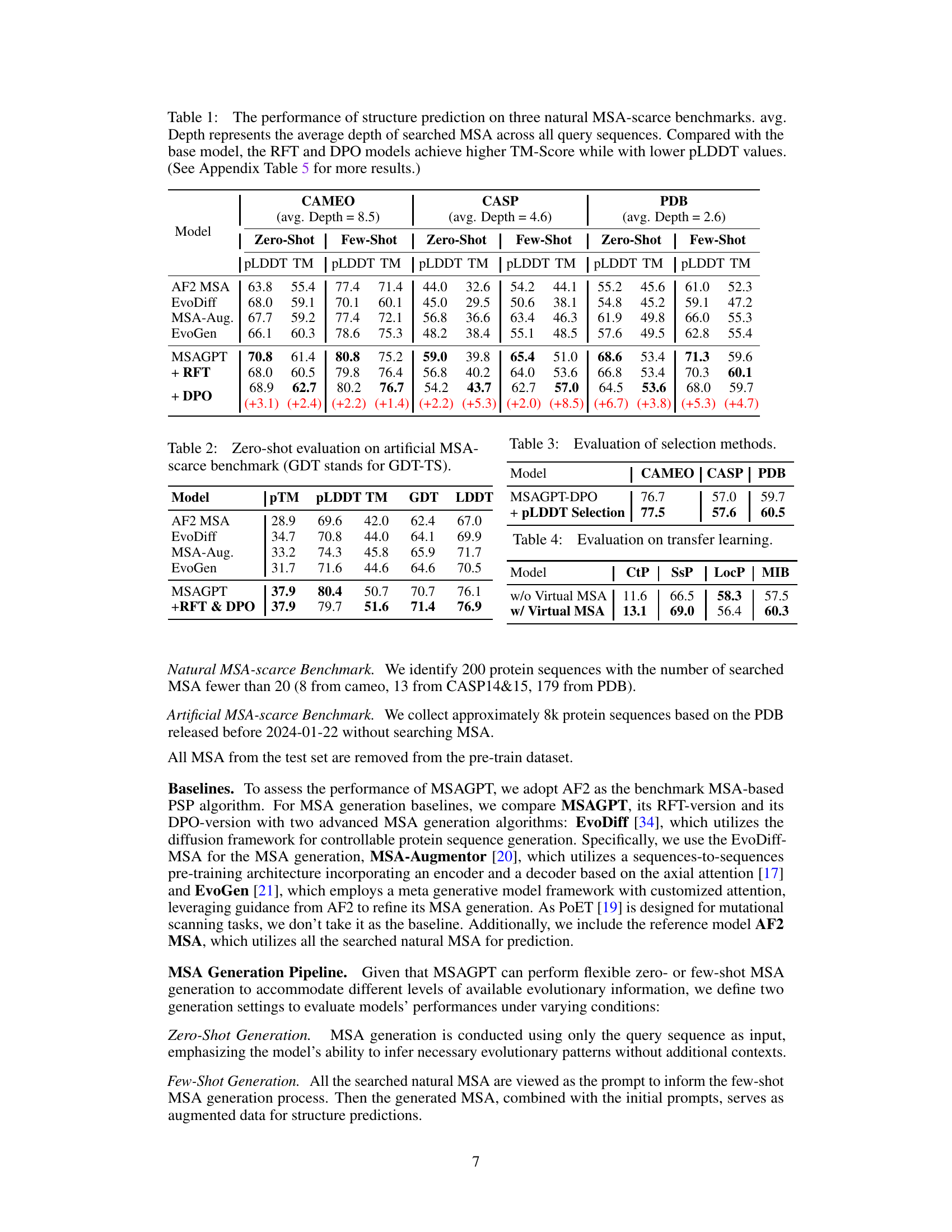
Table 1: The performance of structure prediction on three natural MSA-scarce benchmarks. avg. Depth represents the average depth of searched MSA across all query sequences. Compared with the base model, the RFT and DPO models achieve higher TM-Score while with lower pLDDT values. (See Appendix Table 5 for more results.)
In-depth insights#
MSA Pre-training#
The concept of “MSA Pre-training” in the context of protein structure prediction is a significant advancement. It leverages the power of multiple sequence alignment (MSA) data to pre-train a model capable of generating accurate and informative MSAs, even in scenarios where high-quality MSA data is scarce. This pre-training phase is crucial because it allows the model to learn the intricate co-evolutionary patterns within MSAs, enabling it to predict the accurate spatial arrangements of amino acids. This approach of pre-training addresses a key limitation in current methods, which often struggle to generate virtual MSAs that effectively capture the complex evolutionary relationships among protein sequences. The success of this technique relies on using a 2D positional encoding scheme, capable of capturing both row-wise and column-wise evolutionary information in MSAs more effectively than traditional methods. By using this pre-trained model, the subsequent protein structure prediction task can be achieved via either a zero-shot or few-shot learning approach. This results in improved accuracy, especially when natural MSA data is limited.
2D Positional Encoding#
The concept of “2D Positional Encoding” is a crucial innovation for handling multiple sequence alignments (MSAs) in protein structure prediction. Traditional 1D positional encodings fail to capture the rich, two-dimensional relationships inherent in MSAs where rows represent sequences and columns represent amino acid positions. A 2D encoding scheme simultaneously models both row-wise and column-wise co-evolutionary patterns, which is vital for accurately predicting protein structures. This approach goes beyond simplistic axial attention mechanisms by considering complex interactions across all amino acid sites. By effectively representing this complex interplay using a 2D scheme, the model can better capture the intricate relationships crucial for accurate predictions. This is a significant advance over previous methods that often relied on less efficient, decoupled attention mechanisms that lacked the same level of comprehensive contextual understanding. The effectiveness of this 2D encoding strategy is a key aspect of the model’s overall success. The method significantly enhances the model’s generative capabilities and is a foundational element in the method’s ability to handle the challenging scenarios presented by MSA scarce proteins.
AlphaFold2 Feedback#
The heading ‘AlphaFold2 Feedback’ suggests a crucial refinement stage in the MSAGPT model. It implies that the system uses AlphaFold2’s predictions not just as a final evaluation metric, but as an integral part of the training process. This feedback loop likely enhances the model’s ability to generate more realistic and accurate Multiple Sequence Alignments (MSAs) by penalizing deviations from AlphaFold2’s structural insights. This iterative process is key, because simply generating MSAs and then evaluating them with AlphaFold2 is a passive approach, whereas the feedback mechanism actively guides MSA generation towards biologically plausible outputs. The effectiveness of this method underscores the power of integrating different AI models for improved performance. The use of AlphaFold2’s insights as feedback, rather than a final evaluation, highlights an innovative strategy that likely leads to better MSA generation, especially in situations with scarce homologous sequence data. This approach allows MSAGPT to learn from a highly accurate model, overcoming some of the limitations inherent in purely data-driven MSA generation techniques. The ‘AlphaFold2 Feedback’ component is, therefore, a significant improvement that contributes substantially to the accuracy and reliability of protein structure predictions.
Transfer Learning#
The concept of transfer learning is crucial in the context of protein structure prediction, especially when dealing with limited data. The research explores the potential of utilizing knowledge gained from the pre-training phase on a large MSA dataset to enhance the model’s ability to generate virtual MSA for proteins with scarce homologous information. This transfer learning approach is particularly valuable because it effectively addresses the ‘orphan protein’ problem, where insufficient data hinders accurate structure prediction. The results demonstrate successful transfer learning, showcasing improvement in performance across different protein tasks. This highlights the inherent versatility of the model and its potential applicability beyond primary structure prediction. It opens doors for future research into leveraging this approach for other complex biological tasks by incorporating generated MSA, proving the model’s adaptability. The model’s capacity to generalize and adapt knowledge acquired during pre-training to novel scenarios is a key success of the transfer learning strategy employed.
Limitations and Future#
The research paper’s limitations section would ideally discuss the scaling behavior of the MSAGPT model, acknowledging that its effectiveness has primarily been shown with a model of a specific size and parameter count. Further research should explore how performance changes with varying model sizes and dataset scales. Another key area for improvement would involve assessing the model’s generalizability across a wider range of tasks. While the initial experiments show promise for multiple protein-related tasks, more comprehensive testing is needed to fully understand its capabilities. The section should also address the potential for misuse, particularly concerning the generation of virtual MSAs that could be used to contaminate databases or mislead other research. Finally, discussing any assumptions made during model development and acknowledging the possible impact of these assumptions on the results is crucial. Addressing these limitations through future work would significantly enhance the reliability and impact of MSAGPT.
More visual insights#
More on figures
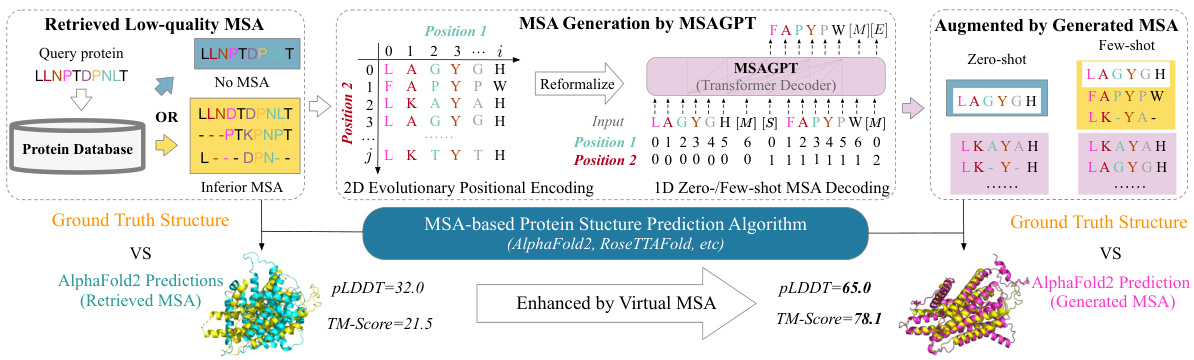
🔼 This figure illustrates the MSAGPT framework. It starts by showing the problem of low-quality or missing MSAs for proteins with limited homologous sequences using conventional methods. Then, it highlights how MSAGPT generates high-quality virtual MSAs using a 2D evolutionary positional encoding and 1D zero-/few-shot MSA decoding. Finally, it shows how these generated MSAs are integrated with an MSA-based protein structure prediction algorithm (like AlphaFold2) to improve the accuracy of structure predictions.
read the caption
Figure 2: The overall framework of prompting protein structure predictions via MSA generation. Left: The challenge faced by conventional search algorithms on protein with scarce homologous sequences, resulting in suboptimal alignments. Middle-to-Right: MSAGPT generates informative and high-quality MSA for such challenging queries, presenting a promising approach to overcoming these limitations. [M] denotes the sequence separator. [S], [E] are the special tokens to represent the start or end of MSA generation.
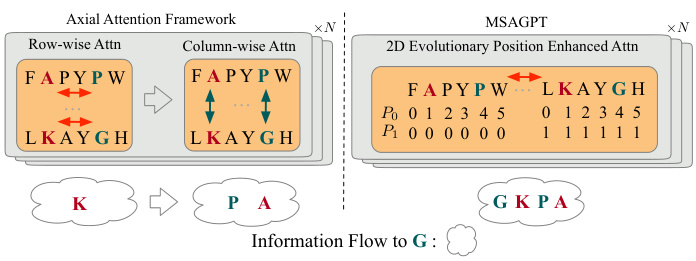
🔼 This figure compares the attention mechanisms used in MSAGPT and axial attention frameworks. It illustrates how MSAGPT’s 2D evolutionary positional encoding allows for more efficient information aggregation compared to the decoupled row-wise and column-wise attentions of axial attention methods. The example focuses on the information flow to amino acid ‘G’, showing how MSAGPT’s approach incorporates both row and column information simultaneously, unlike the two-step approach in axial attention.
read the caption
Figure 3: Comparisons among the axial attention (exemplified by [17]) and the one in MSAGPT in a single layer. Here we focus on the information aggregated to the AA “G”. The 2D evolutionary position enhanced attention shows higher efficiency than the decoupled axial attentions with one-step aggregation to attain sufficient information.
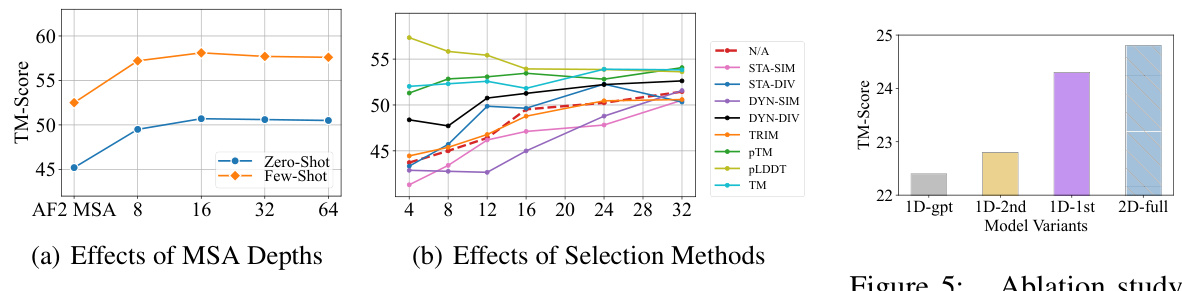
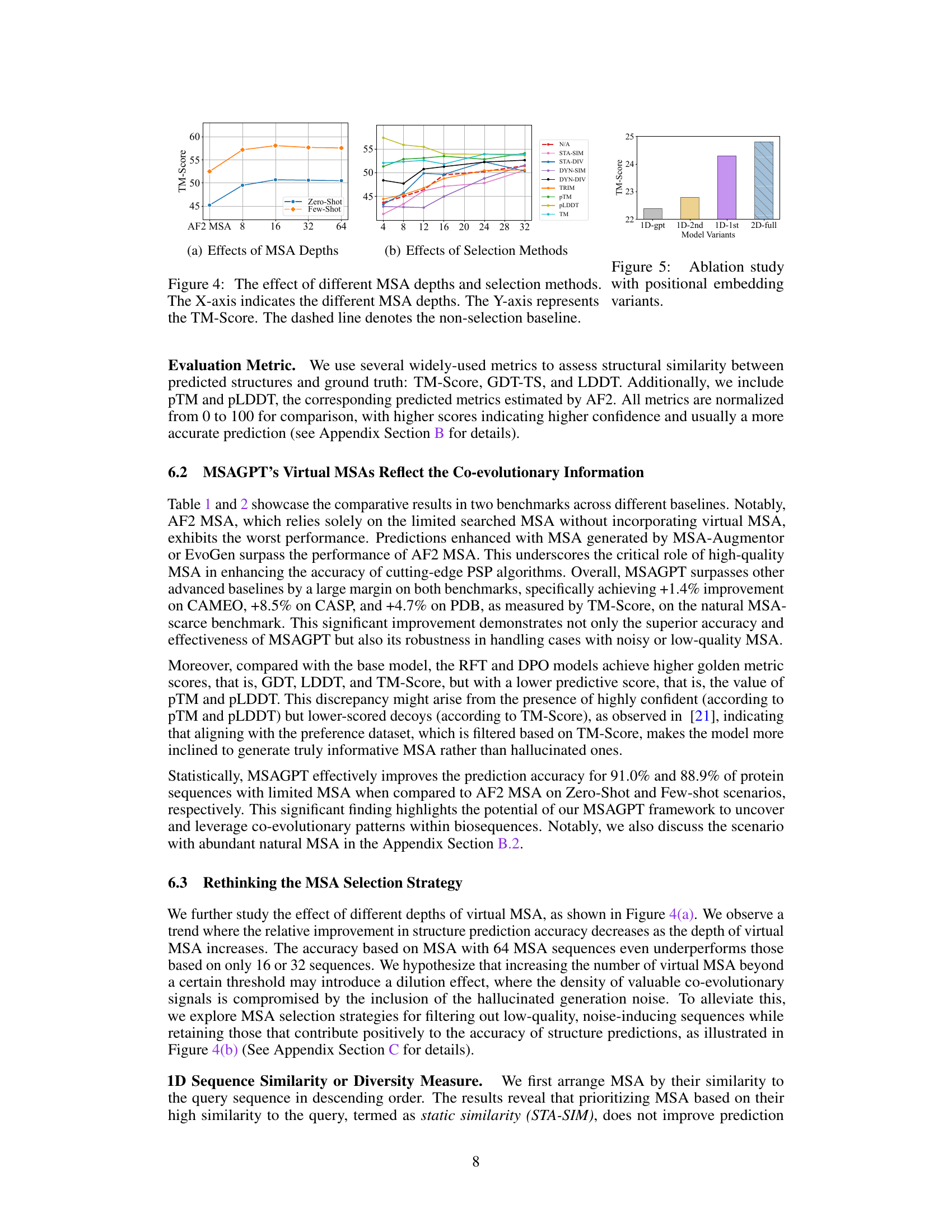
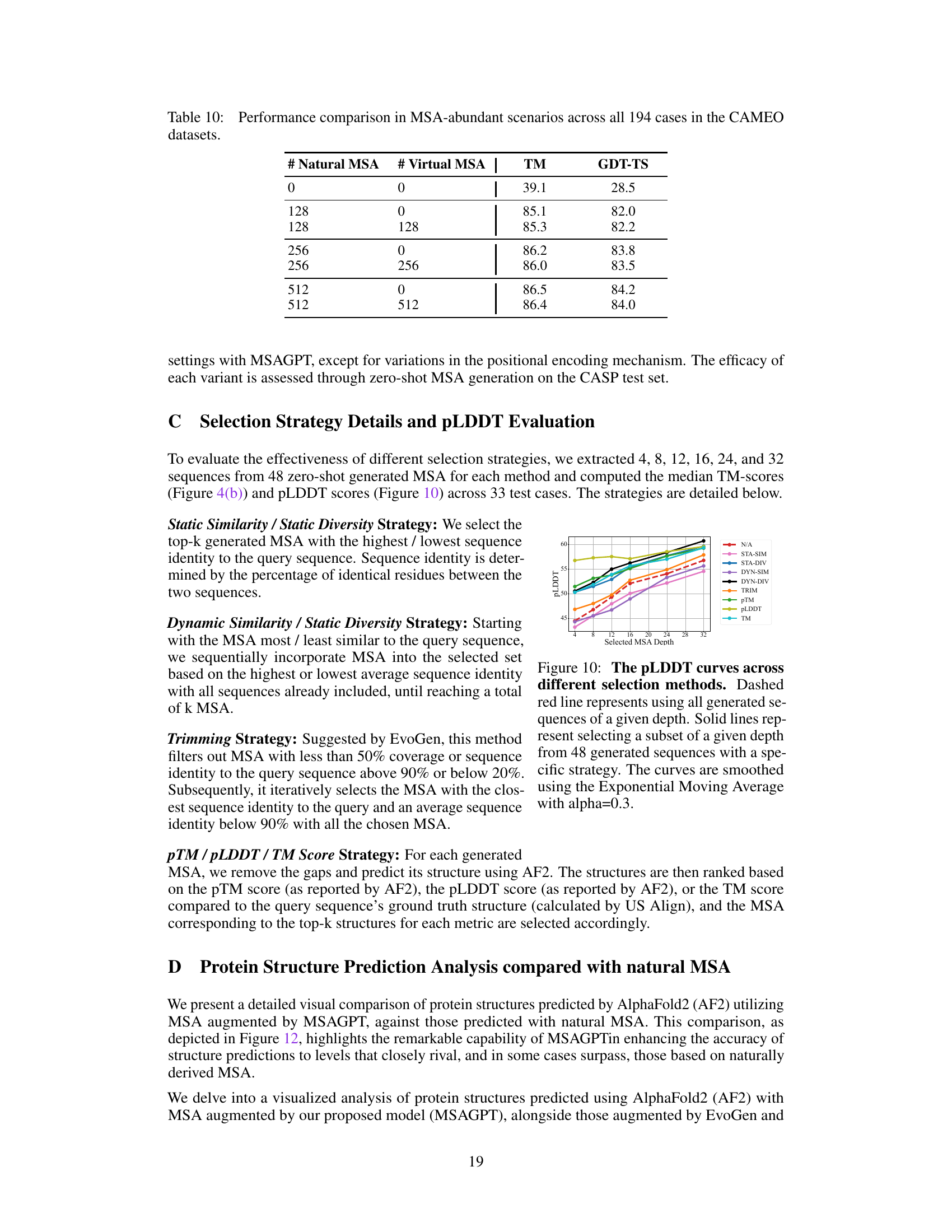
🔼 This figure presents two subfigures showing the effects of MSA depths and selection methods on the TM-Score of protein structure prediction. Subfigure (a) demonstrates that increasing the depth of MSA improves TM-Score up to a certain point after which it starts to decline. Subfigure (b) compares various MSA selection methods, with pLDDT selection showing the best performance.
read the caption
Figure 4: The effect of different MSA depths and selection methods. The X-axis indicates the different MSA depths. The Y-axis represents the TM-Score. The dashed line denotes the non-selection baseline.
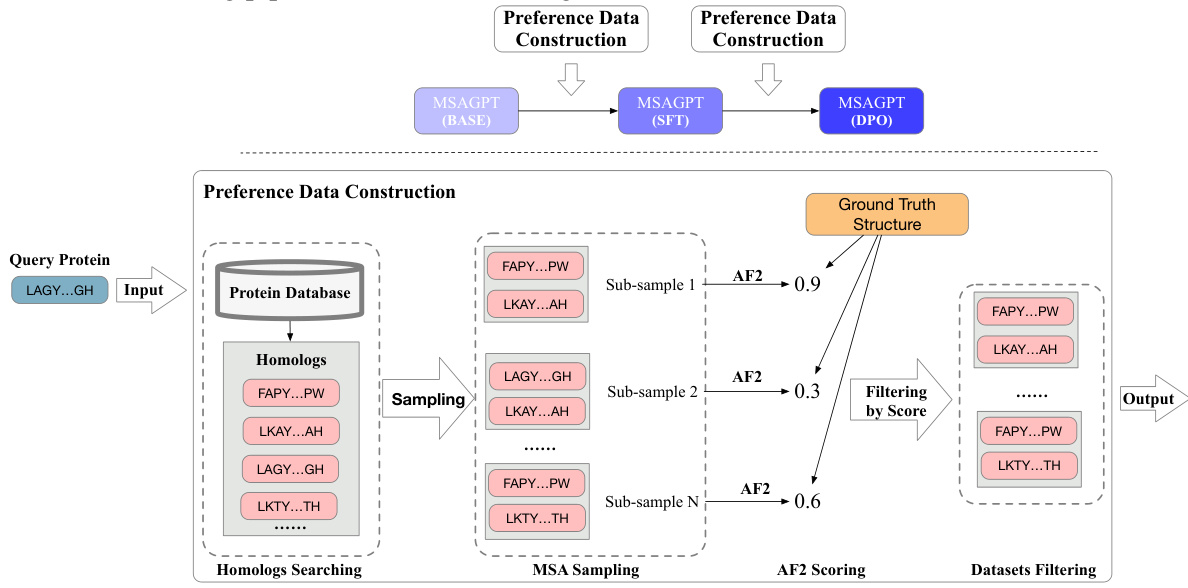
🔼 This figure illustrates the three stages of the MSAGPT training pipeline: MSA generative pre-training, Rejective Fine-tuning (RFT), and Reinforcement Learning from AlphaFold2 Feedback (RLAF). The preference data construction is highlighted, showing how high-quality MSAs are selected based on AlphaFold2 scores. The pipeline begins with MSA generative pre-training using a large dataset. This is followed by RFT, where the model is fine-tuned on a subset of high-quality MSAs selected using AlphaFold2. Finally, RLAF uses AlphaFold2 feedback to further refine the model’s ability to generate informative MSAs. The diagram effectively visualizes the iterative refinement process that leads to improved MSA generation and ultimately better protein structure prediction.
read the caption
Figure 6: The overall training pipeline and the illustration of preference dataset construction process for SFT and DPO learning stages.
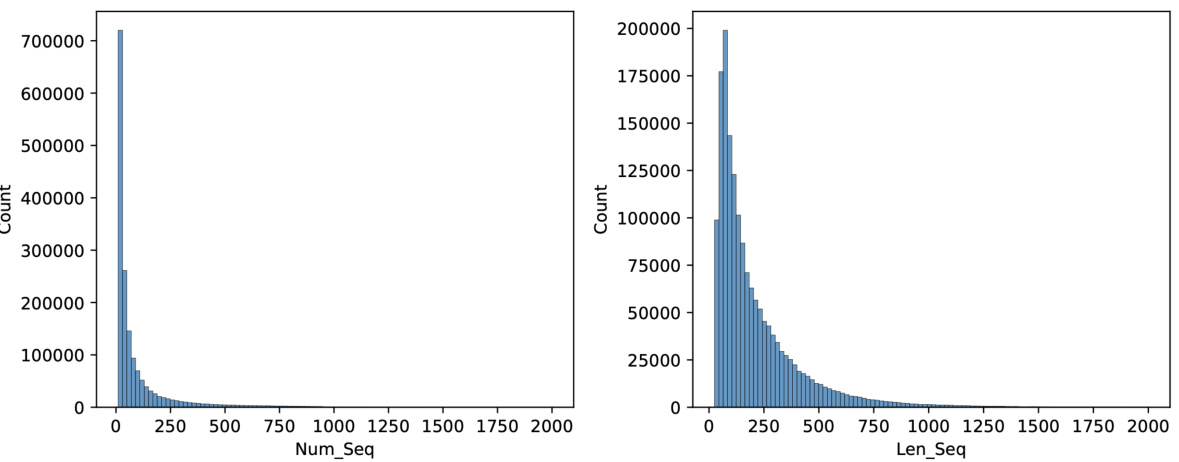
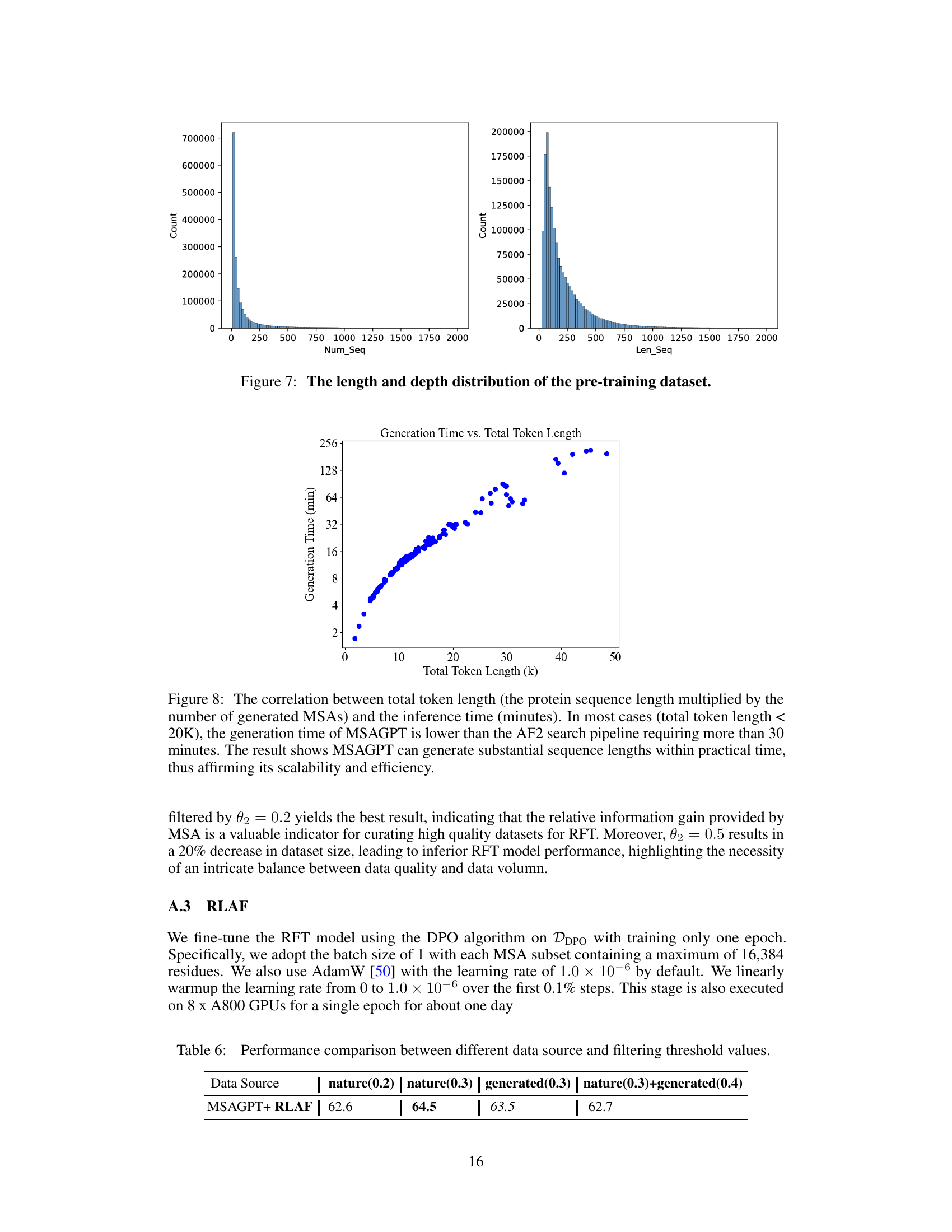
🔼 This figure shows two histograms visualizing the distribution of the pre-training dataset used in MSAGPT. The left histogram displays the number of sequences (Num_Seq) in each MSA, showing a right-skewed distribution with a majority of MSAs having fewer than 200 sequences. The right histogram illustrates the length of sequences (Len_Seq) in the dataset, also showing a right-skewed distribution, with the majority of sequences having lengths less than 500 amino acids. This figure helps understand the characteristics of the dataset used to pre-train MSAGPT and its potential impact on the model’s performance.
read the caption
Figure 7: The length and depth distribution of the pre-training dataset.
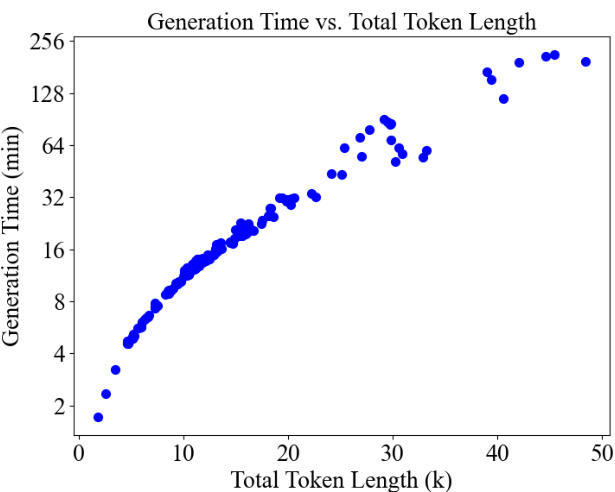
🔼 This figure demonstrates the relationship between the total length of tokens processed by the model and the time taken for generation. It shows that MSAGPT is faster than alternative AlphaFold2 search methods for shorter sequences, highlighting its efficiency and scalability in generating long sequences.
read the caption
Figure 8: The correlation between total token length (the protein sequence length multiplied by the number of generated MSAs) and the inference time (minutes). In most cases (total token length < 20K), the generation time of MSAGPT is lower than the AF2 search pipeline requiring more than 30 minutes. The result shows MSAGPT can generate substantial sequence lengths within practical time, thus affirming its scalability and efficiency.
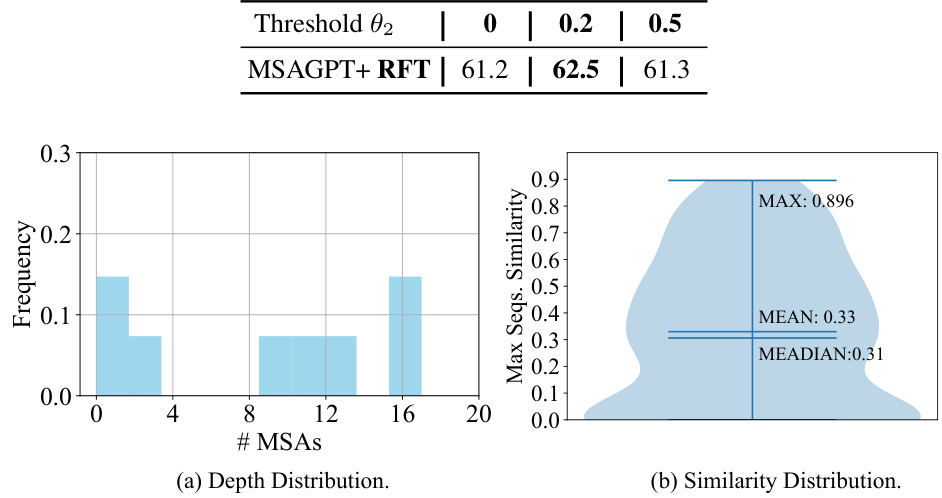
🔼 This figure demonstrates the impact of MSA depth and selection methods on the structure prediction accuracy using MSAGPT. The left subplot shows that increasing the MSA depth to a certain extent improves accuracy, but excessive depth may negatively impact results. The right subplot analyzes various MSA selection strategies, comparing their performance against a baseline with no selection. Different strategies are employed to select MSA based on criteria such as similarity or diversity, with results presented for both few-shot and zero-shot scenarios.
read the caption
Figure 4: The effect of different MSA depths and selection methods. The X-axis indicates the different MSA depths. The Y-axis represents the TM-Score. The dashed line denotes the non-selection baseline.

🔼 This figure displays the performance of different MSA selection strategies on the pLDDT metric. The x-axis represents the number of MSAs selected, and the y-axis represents the pLDDT score. Various selection methods (Static Similarity, Static Diversity, Dynamic Similarity, Dynamic Diversity, Trimming, pTM, pLDDT, TM) are compared to a baseline of using all generated MSAs (dashed red line). The graph shows how the pLDDT score changes as the number of selected MSAs increases for each strategy, illustrating the effectiveness of different approaches in selecting high-quality MSAs.
read the caption
Figure 10: The pLDDT curves across different selection methods. Dashed red line represents using all generated sequences of a given depth. Solid lines represent selecting a subset of a given depth from 48 generated sequences with a specific strategy. The curves are smoothed using the Exponential Moving Average with alpha=0.3.
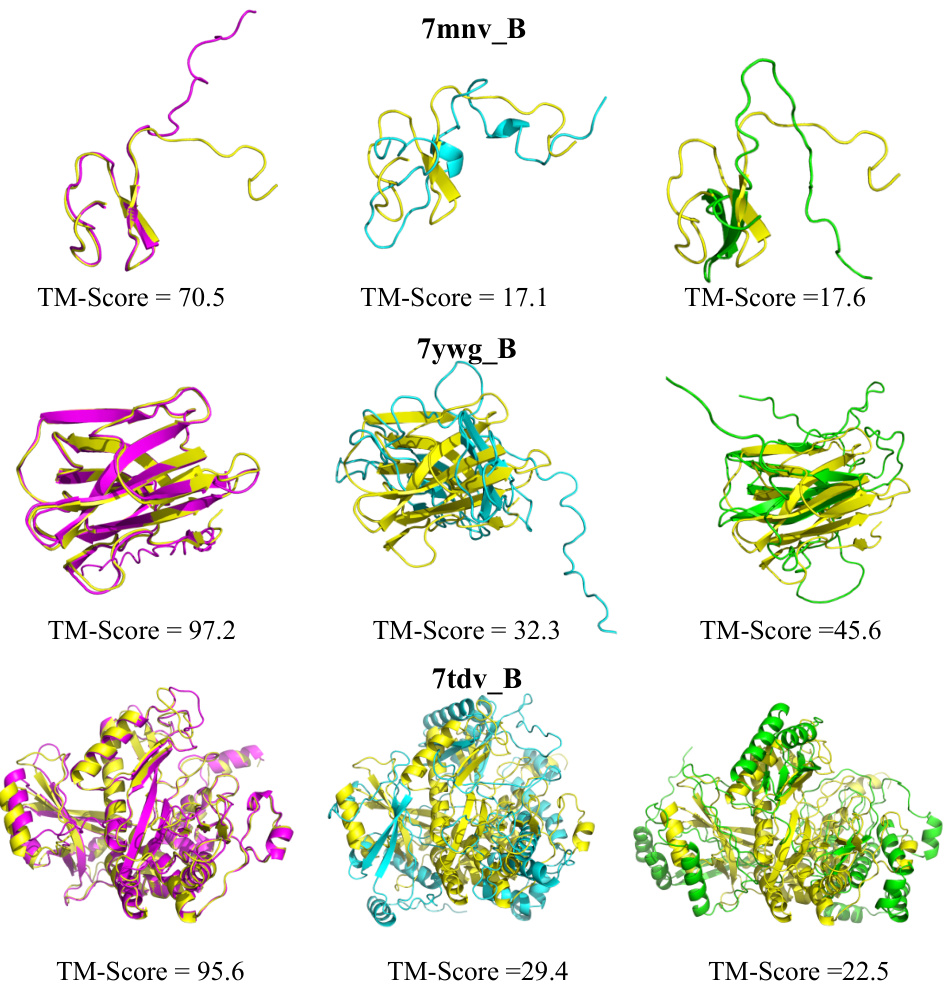
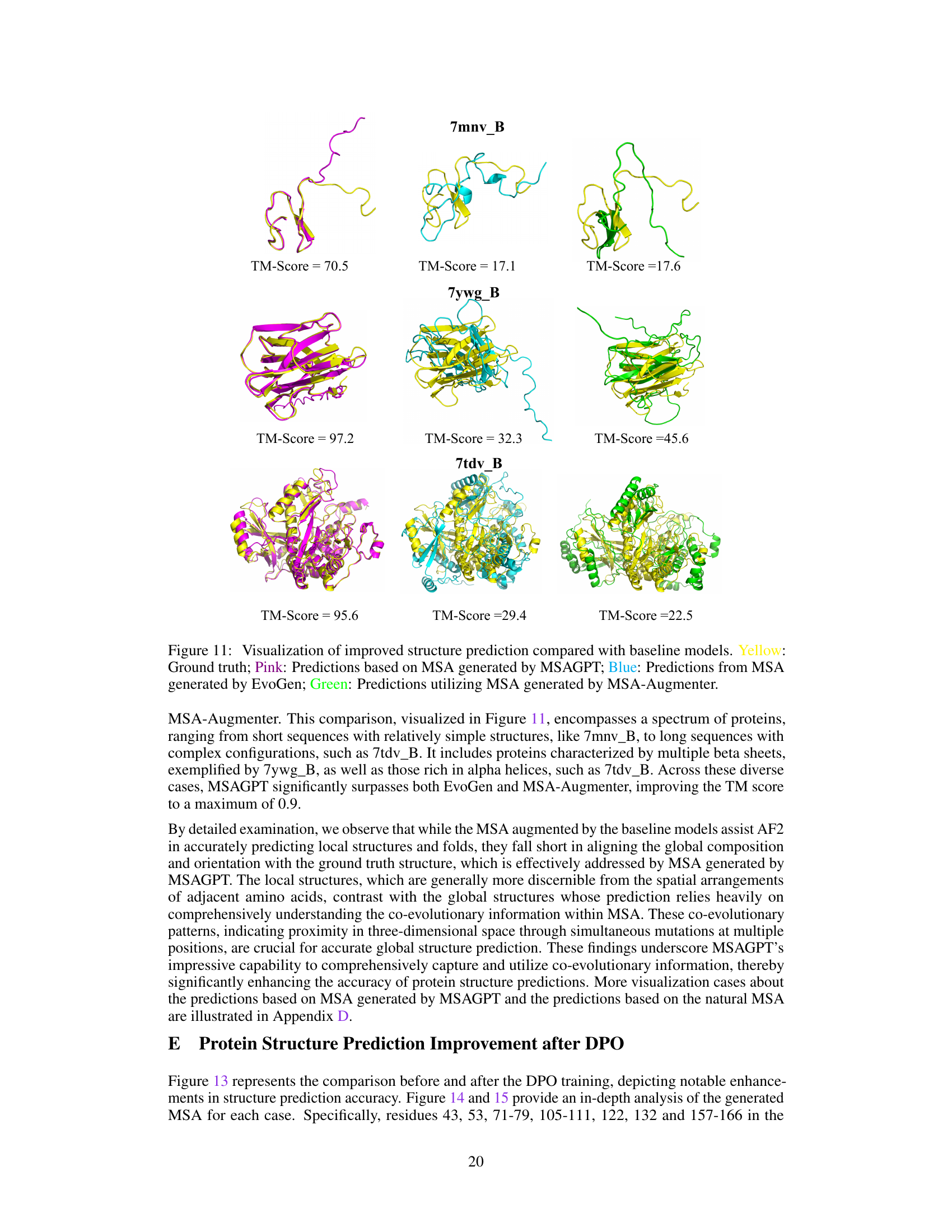
🔼 This figure visually compares the protein structure prediction results of MSAGPT against three baseline methods (EvoGen, MSA-Augmenter, and AF2 using natural MSA). It showcases the improved accuracy of MSAGPT across diverse protein structures, highlighting its ability to accurately predict both local and global structural features. The color-coding helps distinguish between the ground truth structure (yellow), MSAGPT predictions (pink), EvoGen predictions (blue), and MSA-Augmenter predictions (green). The TM-score is provided for each prediction to quantify the level of similarity to the ground truth structure.
read the caption
Figure 11: Visualization of improved structure prediction compared with baseline models. Yellow: Ground truth; Pink: Predictions based on MSA generated by MSAGPT; Blue: Predictions from MSA generated by EvoGen; Green: Predictions utilizing MSA generated by MSA-Augmenter.
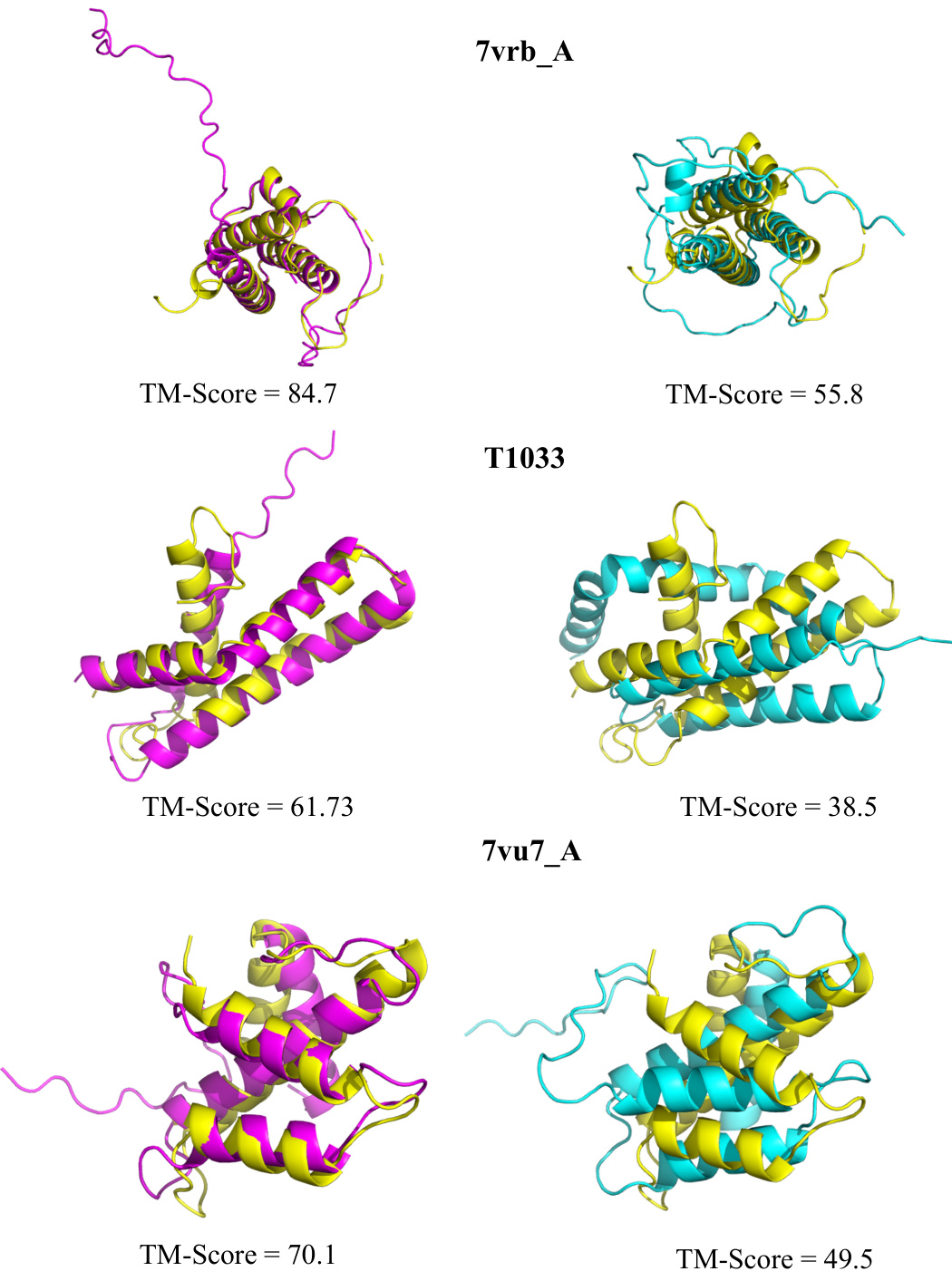
🔼 This figure compares the protein structure prediction results of MSAGPT against several baseline methods (EvoGen and MSA-Augmenter). It uses three different colored structures to represent the ground truth structure, the structure predicted using MSAs generated by MSAGPT, and structures predicted using MSAs from EvoGen and MSA-Augmenter, respectively. The TM-score for each prediction is shown, illustrating the improved accuracy achieved by MSAGPT.
read the caption
Figure 11: Visualization of improved structure prediction compared with baseline models. Yellow: Ground truth; Pink: Predictions based on MSA generated by MSAGPT; Blue: Predictions from MSA generated by EvoGen; Green: Predictions utilizing MSA generated by MSA-Augmenter.
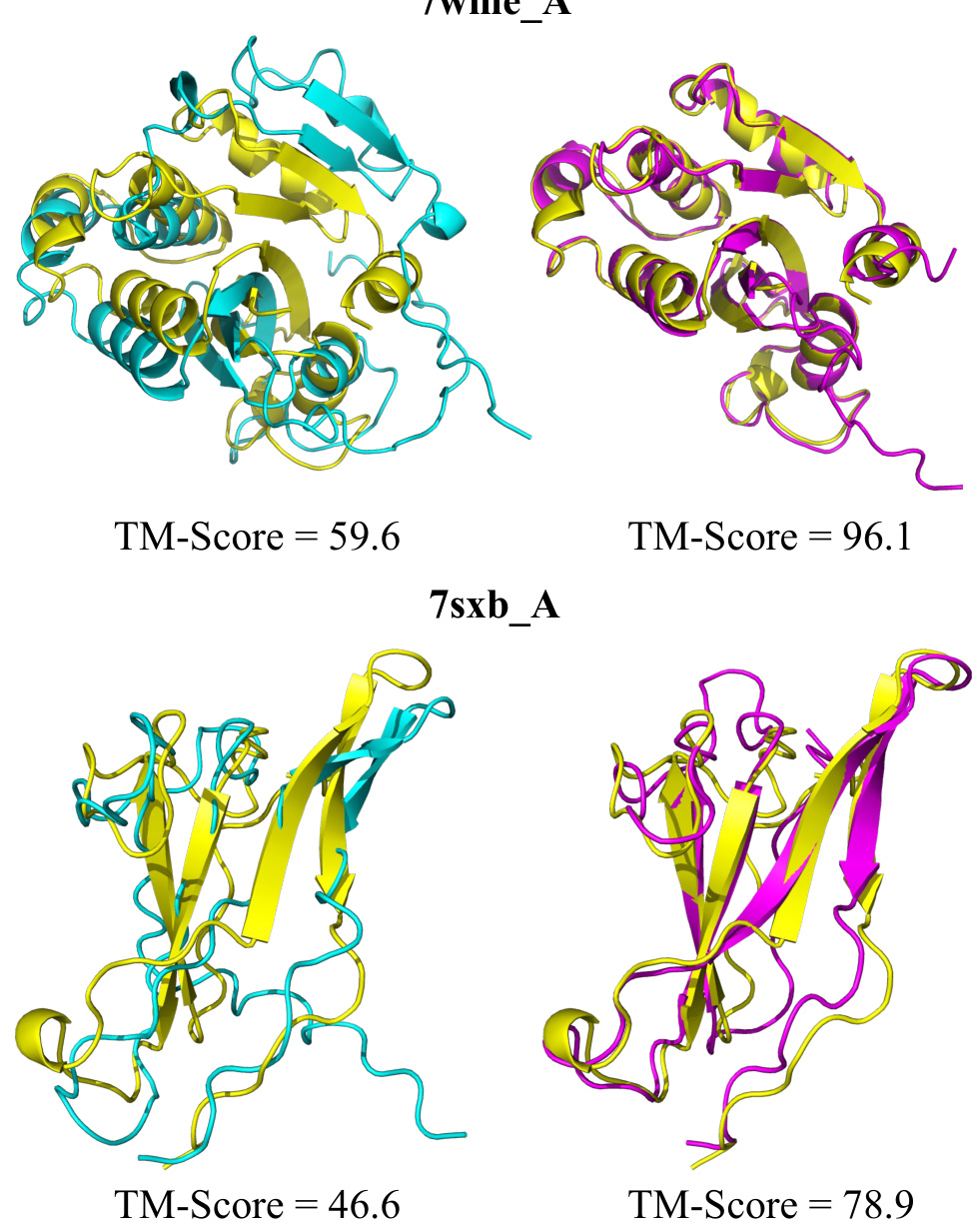
🔼 This figure shows the improved protein structure prediction results after applying the DPO (Direct Preference Optimization) method. The DPO method is a reinforcement learning technique that leverages feedback from AlphaFold2. The figure compares protein structures predicted using MSAGPT alone (blue), MSAGPT with DPO (pink), and the ground truth (yellow). The improved accuracy of the structure predictions after DPO is evident in the close similarity between the pink (MSAGPT with DPO) and yellow (ground truth) structures.
read the caption
Figure 13: Visualization of improved structure prediction after DPO. Yellow: Ground truth; Blue: Predictions based on MSA generated by MSAGPT; Pink: Predictions based on MSA generated by MSAGPT-DPO.
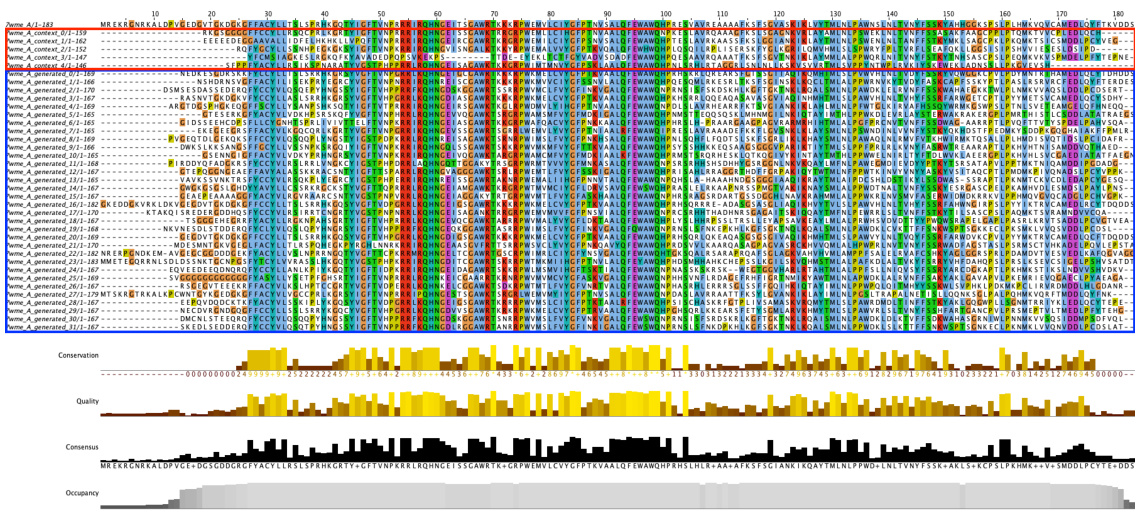
🔼 This figure shows the residue distribution of generated MSA for protein 7wme_A using MSAGPT and MSAGPT with DPO. The top half of the figure displays the MSA generated by MSAGPT. The bottom half shows the MSA generated by MSAGPT with DPO. The red boxes highlight the natural MSA used as input prompts. The blue boxes highlight the generated MSA. The color scheme (clustal by Jalview) indicates the level of conservation at each residue position. The figure visually demonstrates how the use of DPO improves MSA generation quality by comparing the distribution of residues in the MSAs generated by each method.
read the caption
Figure 14: Residue Distribution of Generated MSA for 7wme_A. The red box indicates natural MSA used as prompts during generation. The blue box indicates generated MSA. Residues are colored using the clustal scheme by Jalview.
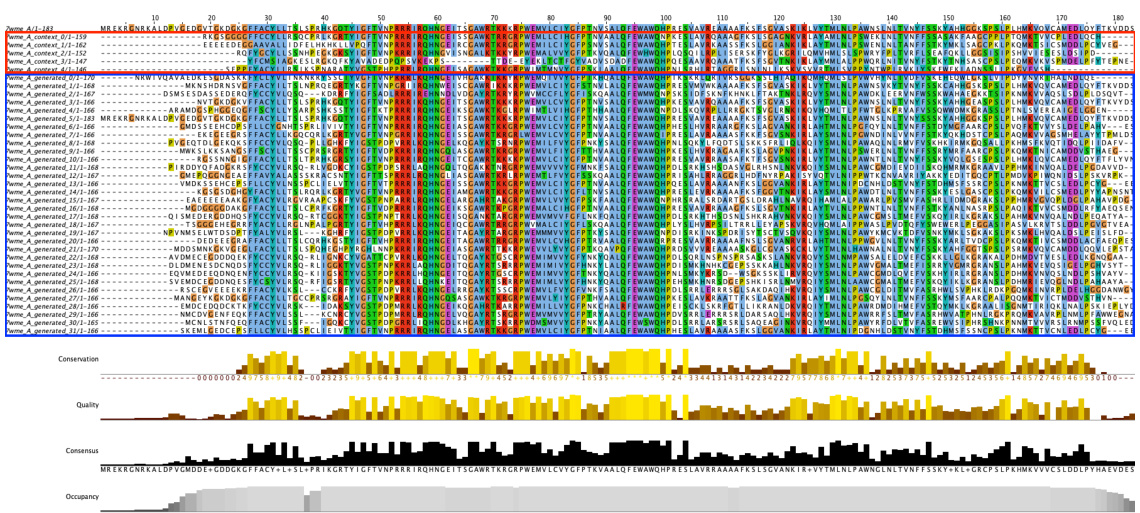
🔼 This figure shows the generated multiple sequence alignment (MSA) by MSAGPT and MSAGPT with DPO for protein 7wme_A. The red box highlights the initial, natural MSA used as input to the model, while the blue box shows the generated MSA. The bottom sections display conservation scores, quality scores, consensus sequences, and occupancy across the alignment, providing a visual representation of sequence similarity and variation within the generated MSA.
read the caption
Figure 14: Residue Distribution of Generated MSA for 7wme_A. The red box indicates natural MSA used as prompts during generation. The blue box indicates generated MSA. Residues are colored using the clustal scheme by Jalview.
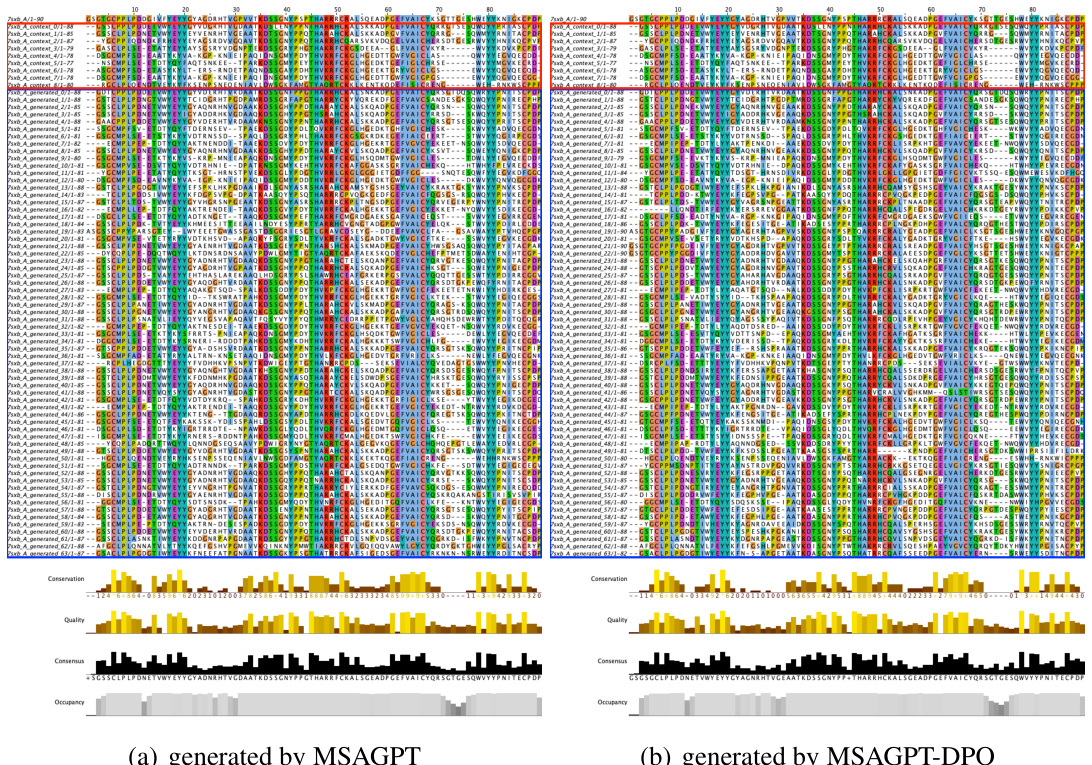
🔼 This figure shows the residue distribution of generated MSAs for protein 7wme_A, comparing the results from MSAGPT and MSAGPT-DPO. The red boxes highlight the natural MSAs used as prompts, while the blue boxes show the generated MSAs. The color scheme (clustal by Jalview) helps visualize the similarities and differences in the amino acid sequences generated by the two models. This visualization is used to demonstrate the model’s ability to generate informative and high-quality MSAs, crucial for accurate protein structure prediction. The difference between the two models likely reflects the effect of the DPO (Direct Preference Optimization) fine-tuning.
read the caption
Figure 14: Residue Distribution of Generated MSA for 7wme_A. The red box indicates natural MSA used as prompts during generation. The blue box indicates generated MSA. Residues are colored using the clustal scheme by Jalview.
More on tables
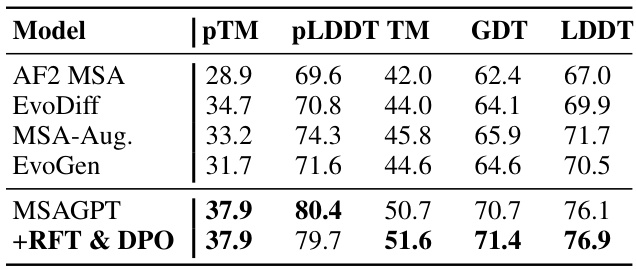
🔼 This table presents the performance of various models on an artificial MSA-scarce benchmark in a zero-shot setting. The benchmark is designed to evaluate the models’ ability to predict protein structures with limited homologous information. The table shows the performance metrics for different models, such as pTM, PLDDT, TM, GDT, and LDDT. These metrics evaluate various aspects of the structure prediction, including the accuracy and confidence of the predictions.
read the caption
Table 2: Zero-shot evaluation on artificial MSA-scarce benchmark (GDT stands for GDT-TS).
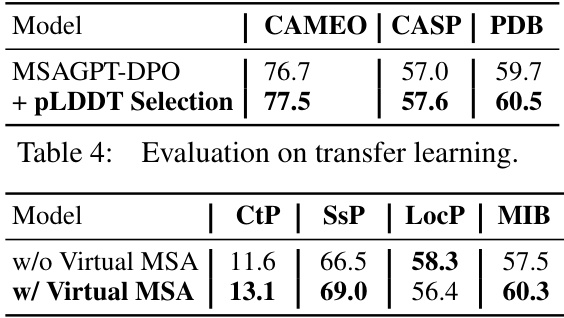
🔼 This table presents the performance comparison of different MSA selection methods used in MSAGPT on three benchmark datasets: CAMEO, CASP, and PDB. It shows how the choice of MSA selection strategy impacts the final TM-Score, reflecting the quality and informativeness of the virtual MSAs generated for protein structure prediction. The results highlight the effectiveness of certain selection strategies in improving prediction accuracy compared to others.
read the caption
Table 3: Evaluation of selection methods.
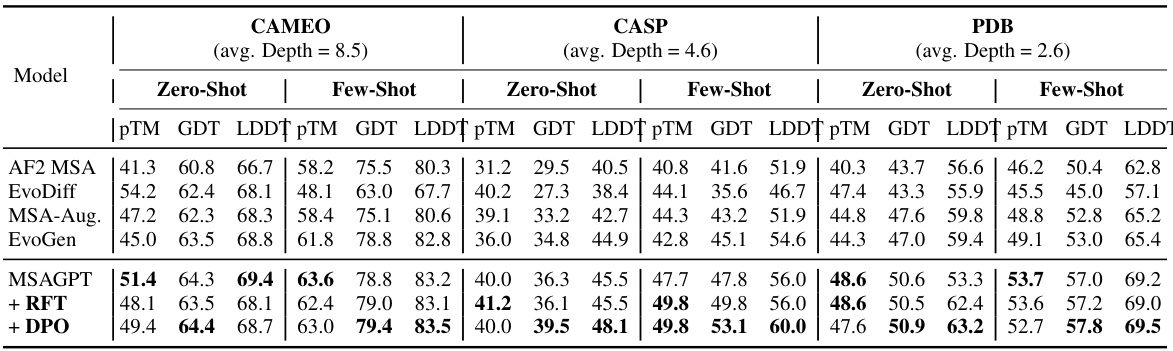
🔼 This table presents the performance comparison of different methods for protein structure prediction on three benchmark datasets with scarce Multiple Sequence Alignment (MSA) data. The performance is evaluated using several metrics including PLDDT, TM-Score, and the average depth of the MSAs. The table shows that MSAGPT, particularly after Rejective Fine-tuning (RFT) and Direct Preference Optimization (DPO), achieves significant improvement in TM-Score compared to other methods, while maintaining relatively good pLDDT scores.
read the caption
Table 1: The performance of structure prediction on three natural MSA-scarce benchmarks. avg. Depth represents the average depth of searched MSA across all query sequences. Compared with the base model, the RFT and DPO models achieve higher TM-Score while with lower pLDDT values. (See Appendix Table 5 for more results.)

🔼 This table presents the performance of protein structure prediction using different methods on three benchmark datasets with scarce Multiple Sequence Alignment (MSA) data. It compares the performance of AlphaFold2 (AF2) with different MSA sources and methods, including AlphaFold2 using the original MSA, EvoDiff, MSA-Augmentor, EvoGen, and MSAGPT (with and without Rejective Fine-Tuning (RFT) and Direct Preference Optimization (DPO)). The metrics used to evaluate the performance are PLDDT (predicted Local Distance Difference Test), TM-score (Template Modeling Score), and the average depth of MSA used.
read the caption
Table 1: The performance of structure prediction on three natural MSA-scarce benchmarks. avg. Depth represents the average depth of searched MSA across all query sequences. Compared with the base model, the RFT and DPO models achieve higher TM-Score while with lower pLDDT values. (See Appendix Table 5 for more results.)
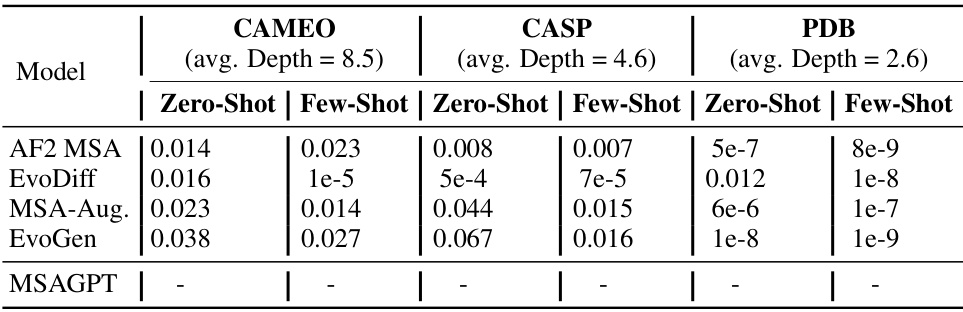
🔼 This table presents the p-values from paired Student’s t-tests comparing the TM-scores of MSAGPT against other methods across three benchmark datasets (CAMEO, CASP, and PDB). The tests assess the statistical significance of the differences in TM-scores between MSAGPT and each competing method under zero-shot and few-shot scenarios. A p-value less than 0.05 suggests a statistically significant difference in performance.
read the caption
Table 8: The paired Student's t-test between MSAGPT and other baselines on three benchmarks based on the TM-Score, where the p-value less than 0.05 indicates the result is said to be statistically significant.
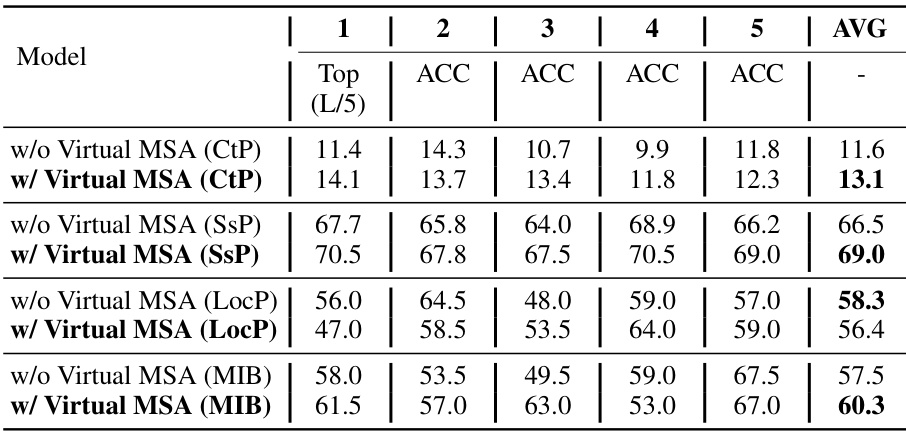
🔼 This table presents the results of a 5-fold cross-validation experiment evaluating the impact of virtual MSAs generated by MSAGPT on four protein-related tasks: Contact Prediction (CtP), Secondary Structure Prediction (SSP), Localization Prediction (LocP), and Metal Ion Binding (MIB). For each task, the table shows the performance (Top1 accuracy and average accuracy across the 5 folds) both with and without the inclusion of virtual MSAs generated by MSAGPT. The comparison highlights the potential benefits of using MSAGPT-generated virtual MSAs to improve the performance of protein-related tasks beyond structure prediction.
read the caption
Table 9: The results of 5-fold cross-validation performance between with or without virtual MSA generated by MSAGPT on four protein-related tasks.
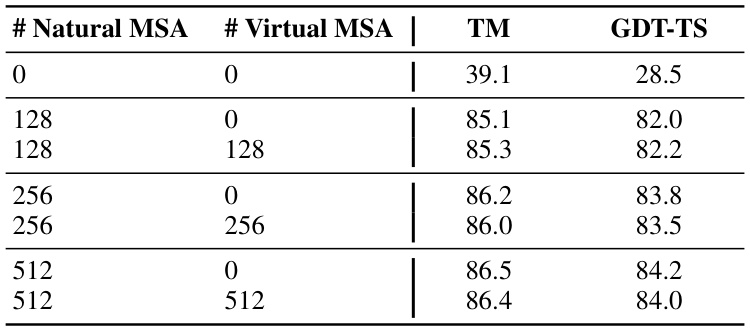
🔼 This table presents a performance comparison of structure prediction in scenarios with abundant MSAs. It compares results using different combinations of natural and virtual MSAs generated by MSAGPT. The results are measured by TM score and GDT-TS, both of which evaluate the similarity between the predicted and ground-truth protein structures. The purpose is to demonstrate that adding virtual MSAs from MSAGPT does not significantly improve performance when a sufficient number of natural MSAs are already available.
read the caption
Table 10: Performance comparison in MSA-abundant scenarios across all 194 cases in the CAMEO datasets.
Full paper#