↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Single-cell ATAC sequencing (scATAC-seq) is a powerful technique for studying chromatin accessibility at a single-cell resolution. However, scATAC-seq data often suffers from sparsity and noise, hindering accurate analysis. Current methods for scATAC-seq data analysis are often task-specific, lacking a unified framework for multiple tasks. This poses significant challenges for researchers seeking comprehensive analysis.
To overcome these limitations, the researchers proposed ATAC-Diff, a novel and versatile framework that uses a latent diffusion model. This model incorporates an auxiliary module to encode latent variables, which helps to improve the quality of generated data and enable more informative downstream analyses. Through extensive experiments, they demonstrated that ATAC-Diff outperforms existing state-of-the-art methods in both data generation and analysis tasks. This work provides a significant advance for the field by offering a unified and powerful framework for scATAC-seq data analysis and simulation.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers working with single-cell ATAC-seq data due to its introduction of ATAC-Diff, a novel and versatile framework for both data generation and analysis, outperforming existing methods. This significantly improves downstream analysis and enables the generation of high-quality synthetic data, addressing the crucial issue of data sparsity and noise in scATAC-seq experiments. Its implications are far-reaching, impacting many fields that utilize single-cell genomics. The model’s flexibility to handle various tasks within a single framework is also a significant advance.
Visual Insights#
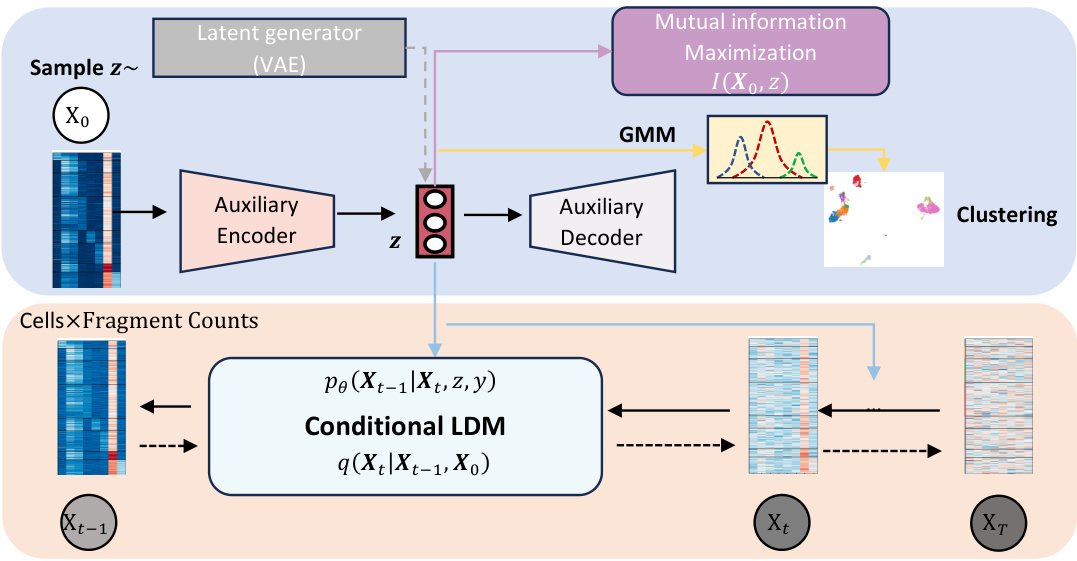
The figure illustrates the ATAC-Diff framework, a versatile informative diffusion model for single-cell ATAC-seq data generation and analysis. It shows how the model uses an auxiliary module to encode input scATAC-seq data into a low-dimensional semantic representation, which is then used to condition a diffusion model for data generation and analysis. The process involves a latent generator (VAE), an auxiliary encoder, a conditional latent diffusion model (LDM), and an auxiliary decoder. Mutual information maximization and a Gaussian Mixture Model (GMM) are also used to improve the model’s performance. The output includes high-quality synthetic scATAC-seq data and cell type identification.
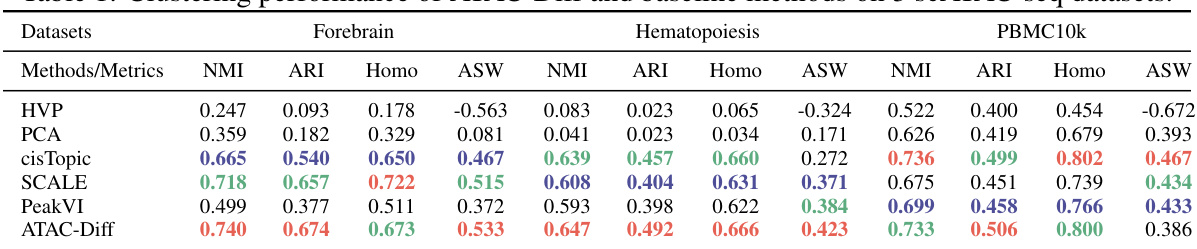
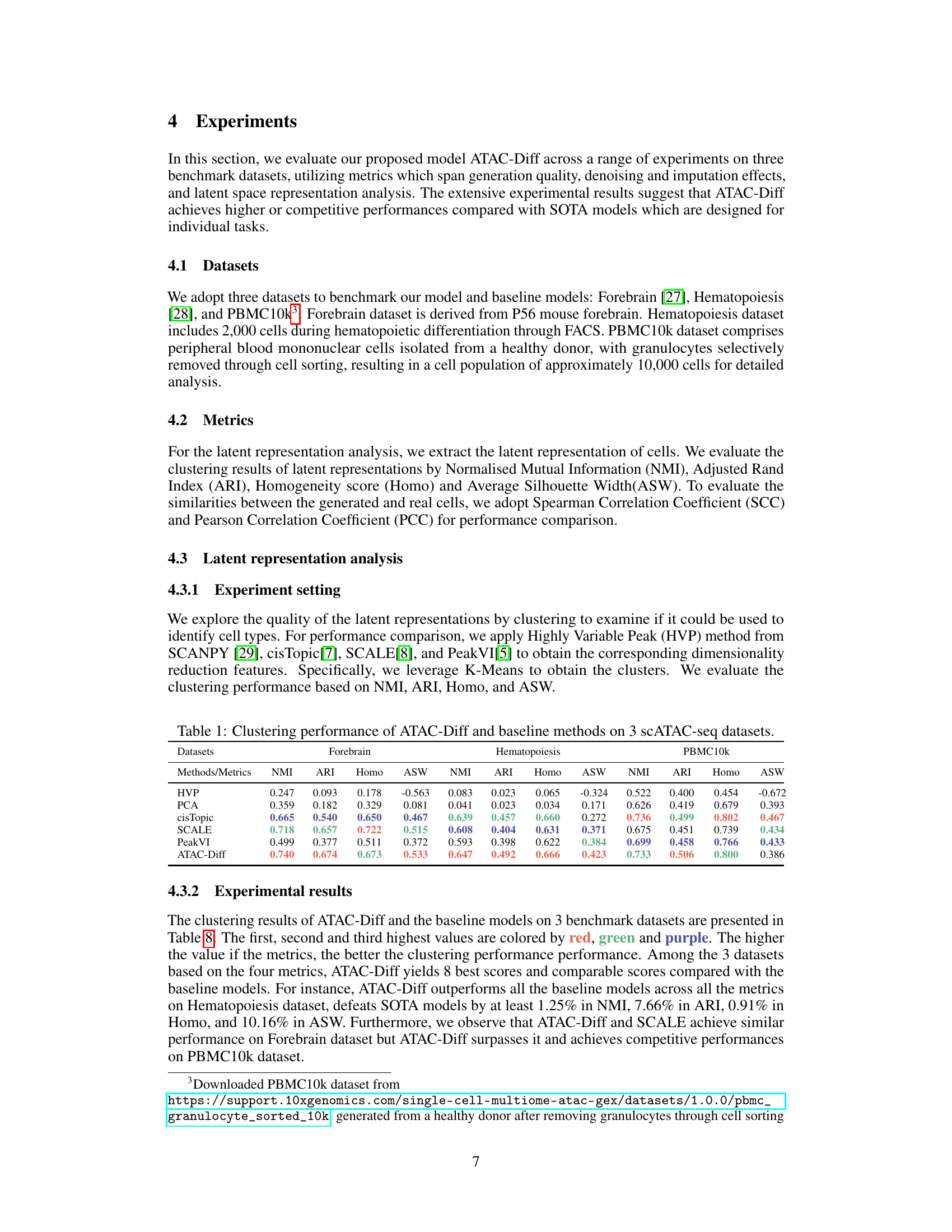
This table presents a comparison of the clustering performance of ATAC-Diff against several baseline methods (HVP, PCA, cisTopic, SCALE, PeakVI) across three different scATAC-seq datasets (Forebrain, Hematopoiesis, PBMC10k). The performance is evaluated using four metrics: Normalized Mutual Information (NMI), Adjusted Rand Index (ARI), Homogeneity (Homo), and Average Silhouette Width (ASW). Higher values generally indicate better clustering performance.
In-depth insights#
ATAC-Diff Overview#
ATAC-Diff is a novel versatile framework designed for single-cell ATAC-seq data generation and analysis. It leverages a latent diffusion model conditioned on auxiliary variables to enable the generation of high-quality, biologically realistic data. The model incorporates a Gaussian Mixture Model (GMM) as a prior, allowing it to capture the inherent multimodality of scATAC-seq data. Furthermore, mutual information maximization is used as a regularizer to ensure the model maintains meaningful latent representations. This approach allows ATAC-Diff to outperform state-of-the-art methods in tasks such as data generation, denoising, imputation and clustering analysis, demonstrating its effectiveness across diverse applications in genomics research. The framework’s ability to handle multiple tasks within a unified model is a significant advancement, simplifying workflows and fostering a more comprehensive understanding of chromatin accessibility. The integration of GMM and mutual information is crucial in preventing the model from becoming overly reliant on noise during training and improves the overall quality of results.
Diffusion Model#
Diffusion models are powerful generative models that gradually add noise to data until it becomes pure noise, and then learn to reverse this process to generate new data. The core idea is to learn a Markov chain that transforms data into noise and its reverse. This approach offers several advantages, including high-quality sample generation and the ability to handle various data modalities. In the context of single-cell ATAC-seq data, diffusion models address the challenge of sparsity and noise inherent in the data by learning the underlying data distribution and generating high-quality samples. The conditional nature of diffusion models allows for control over generation process, such as conditioning on cell type or other metadata. This feature is particularly valuable for analyzing scATAC-seq data, where integrating different biological sources of information can provide valuable insights. However, challenges remain in ensuring that learned latent spaces are meaningful and interpretable. The method requires careful consideration of hyperparameter tuning and a thorough understanding of the model’s behavior in generating diverse yet realistic scATAC-seq data.
Auxiliary Module#
The auxiliary module in this ATAC-seq diffusion model, ATAC-Diff, plays a crucial role in bridging the gap between raw genomic data and the latent space of the diffusion model. It acts as a semantic encoder, transforming the raw, high-dimensional scATAC-seq data into a lower-dimensional, meaningful representation. This is achieved by incorporating a Gaussian Mixture Model (GMM) as the prior, which helps capture the multi-modal nature of scATAC-seq data stemming from diverse cell types. Further enhancing its effectiveness is the incorporation of mutual information between the latent variables and the observed data, which acts as a regularizer. This regularizer helps maintain the connection between the latent and observed space, preventing the model from neglecting crucial latent information. The auxiliary module thus provides a refined and informative latent representation suitable for downstream analyses like clustering and visualization, as well as high-quality scATAC-seq data generation in the diffusion process.
Experimental Results#
The “Experimental Results” section would ideally present a thorough evaluation of the ATAC-Diff model, comparing its performance against state-of-the-art methods across diverse tasks such as clustering, data generation, denoising, and imputation. Quantitative metrics like NMI, ARI, ASW, SCC, and PCC should be reported for each task, showcasing ATAC-Diff’s capabilities on various benchmark datasets. Visualizations (e.g., UMAP plots) would effectively demonstrate the model’s ability to separate cell types and capture biological relationships in the latent space. A detailed analysis of the results is crucial, highlighting strengths and limitations, perhaps attributing superior performance to specific design choices (e.g., the auxiliary module or mutual information regularization). Furthermore, the discussion should connect the findings back to the claims presented in the introduction, reinforcing the significance of the ATAC-Diff framework for single-cell ATAC-seq data analysis and generation.
Future Directions#
Future research could explore integrating ATAC-Diff with other omics data, such as single-cell RNA-seq, to gain a more holistic understanding of gene regulation. This multi-modal approach could reveal intricate relationships between chromatin accessibility and gene expression, leading to more precise and comprehensive biological interpretations. Another avenue is developing more sophisticated conditional generation models, allowing for the creation of synthetic scATAC-seq data with specific characteristics tailored to downstream analyses. This would improve data augmentation and simulations for scenarios with limited samples or specific cell types of interest. Finally, enhancing the interpretability of the learned latent space is crucial for uncovering deeper biological insights. Developing techniques to map latent variables to biologically meaningful features would significantly increase the usability and value of the generated data, ultimately enhancing our understanding of cellular processes and regulatory mechanisms.
More visual insights#
More on figures
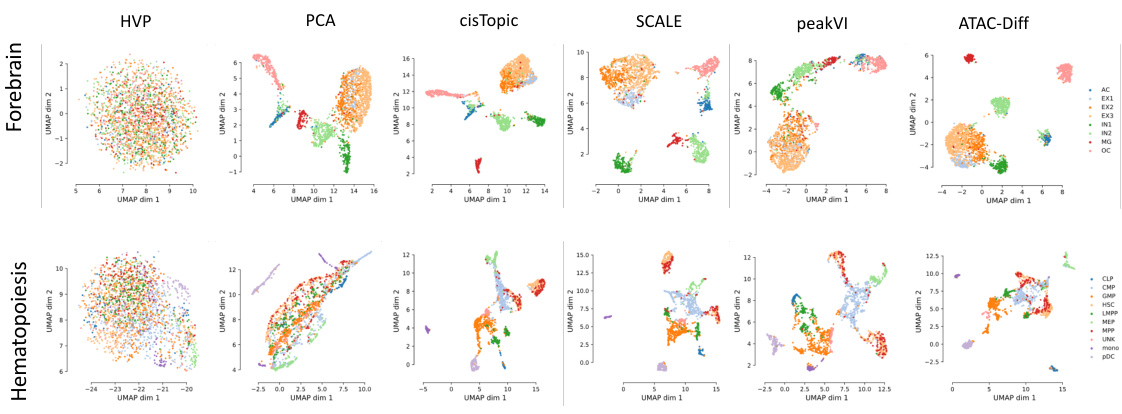
UMAP plots showing the results of dimensionality reduction and clustering on Forebrain and Hematopoiesis datasets using several methods, including ATAC-Diff. Each point represents a cell, colored by cell type. ATAC-Diff shows better separation of cell types compared to other methods.
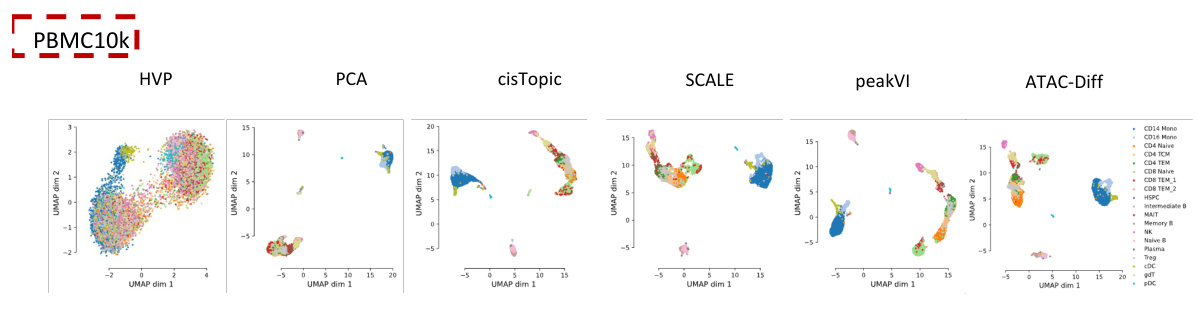
This figure visualizes the results of UMAP dimensionality reduction applied to highly variable peak values and features extracted by different methods (PCA, cisTopic, SCALE, PeakVI, and ATAC-Diff) on two scATAC-seq datasets (Forebrain and Hematopoiesis). It shows how well each method separates different cell types in the low-dimensional UMAP space. The visualization for the PBMC10k dataset is provided in the Appendix.
More on tables
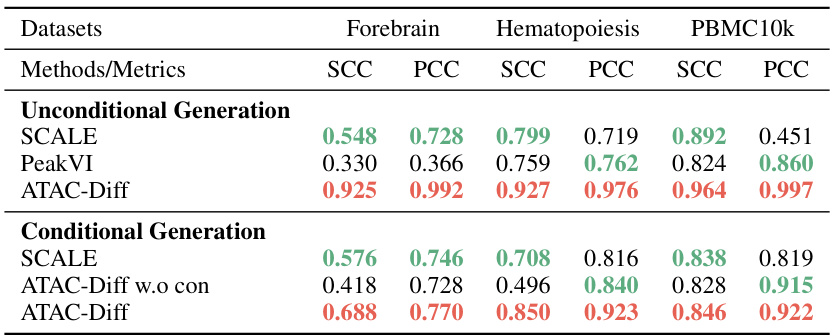
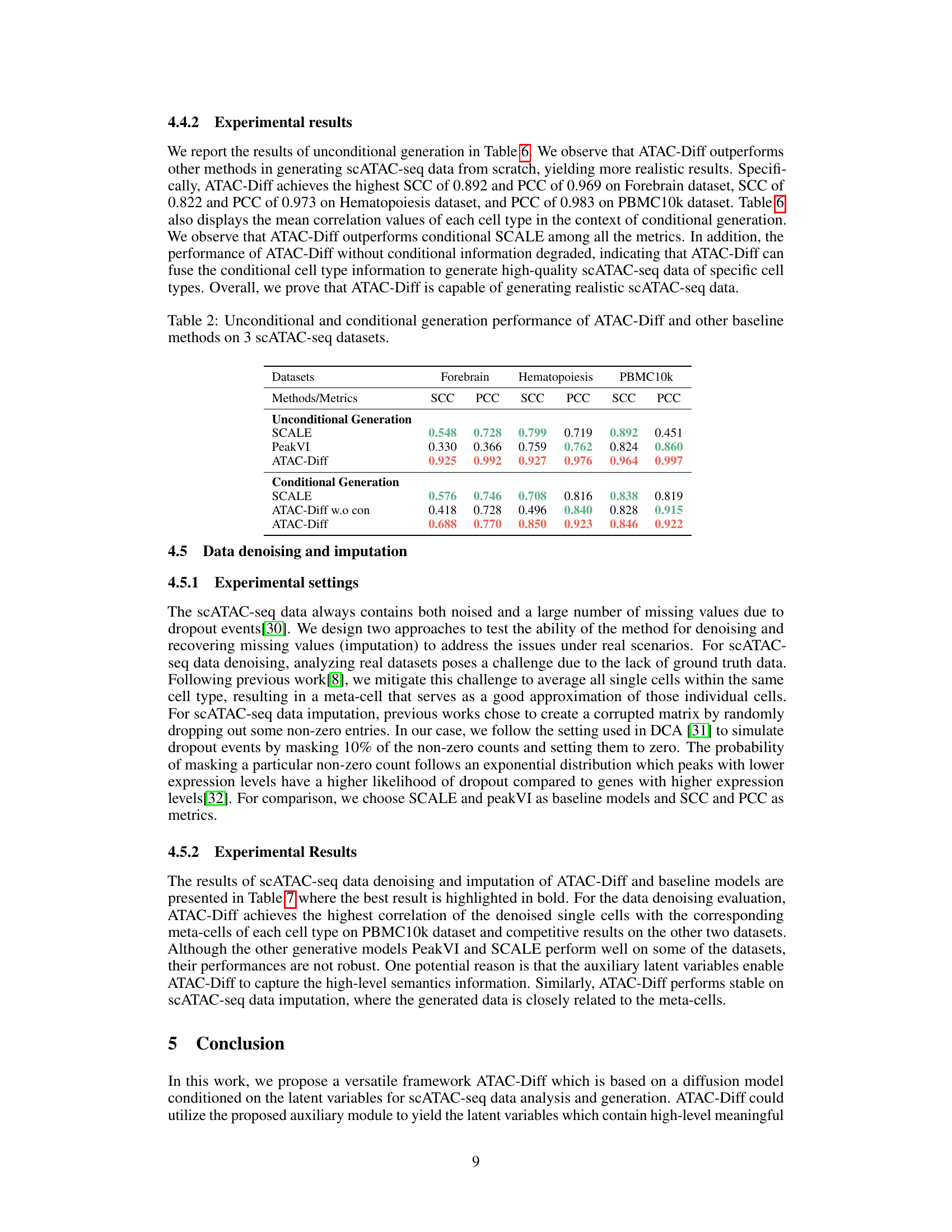
This table presents the results of unconditional and conditional generation experiments performed on three scATAC-seq datasets (Forebrain, Hematopoiesis, and PBMC10k). It compares the performance of ATAC-Diff against baseline models (SCALE and PeakVI) using two metrics: Spearman Correlation Coefficient (SCC) and Pearson Correlation Coefficient (PCC). Both unconditional and conditional (with cell type information) generation scenarios are evaluated to showcase ATAC-Diff’s ability to generate realistic and cell type-specific scATAC-seq data.
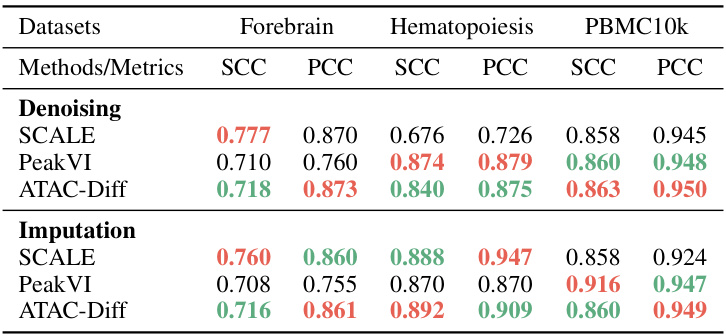
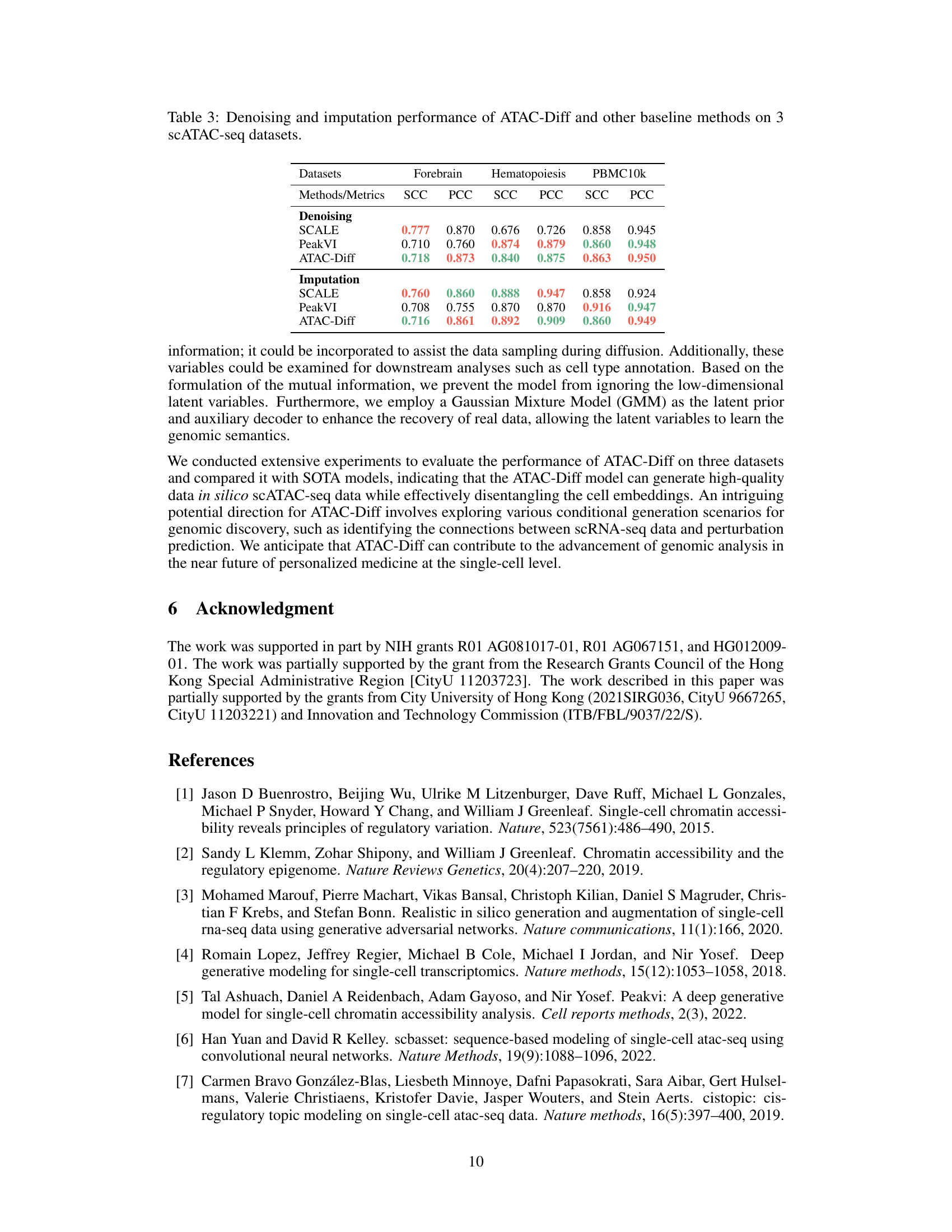
This table presents the results of denoising and imputation experiments performed using ATAC-Diff and several baseline methods across three scATAC-seq datasets (Forebrain, Hematopoiesis, and PBMC10k). The performance is evaluated using two metrics: Spearman Correlation Coefficient (SCC) and Pearson Correlation Coefficient (PCC). Higher SCC and PCC values indicate better performance in denoising and imputation tasks. The table is structured to compare the performance of ATAC-Diff against the baseline methods (SCALE and PeakVI) on each of the three datasets for both denoising and imputation separately.
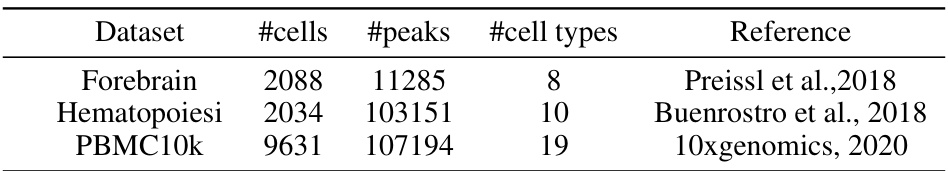
This table summarizes the number of cells, peaks, and cell types for three scATAC-seq datasets used in the paper: Forebrain, Hematopoiesis, and PBMC10k. It also provides references for where the datasets originated.
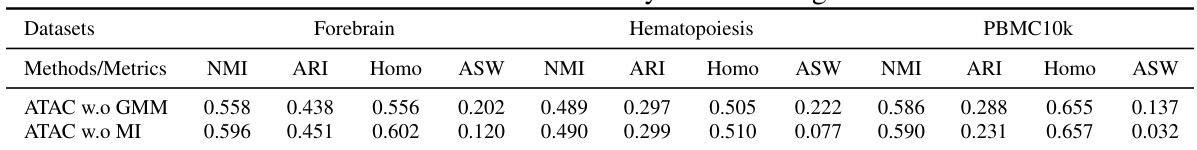
This table presents the clustering performance comparison of ATAC-Diff with and without GMM and MI modules, across three benchmark datasets (Forebrain, Hematopoiesis, and PBMC10k). The metrics used are Normalized Mutual Information (NMI), Adjusted Rand Index (ARI), Homogeneity, and Average Silhouette Width (ASW), which are common metrics for evaluating clustering quality. The results demonstrate the contribution of each component of ATAC-Diff to the clustering performance.
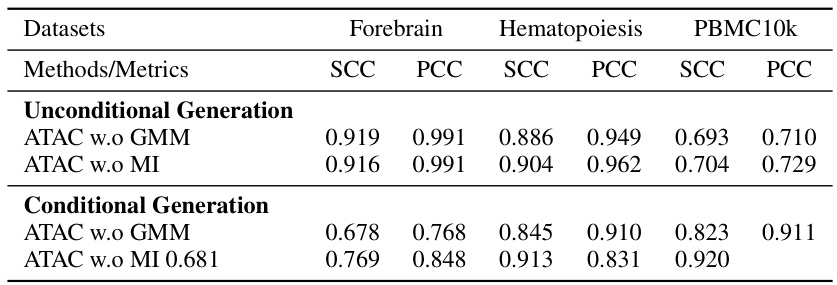
This table shows the results of unconditional and conditional generation experiments on three scATAC-seq datasets (Forebrain, Hematopoiesis, and PBMC10k). It compares the performance of ATAC-Diff against baseline methods (SCALE and ATAC-Diff with either the GMM or Mutual Information (MI) module removed) using two metrics: Spearman Correlation Coefficient (SCC) and Pearson Correlation Coefficient (PCC). Higher SCC and PCC values indicate better performance in generating realistic scATAC-seq data. The results demonstrate the improved performance of ATAC-Diff for both unconditional and conditional data generation tasks, indicating the effectiveness of the model architecture in capturing the underlying data distribution and utilizing conditional information to generate high-quality data.
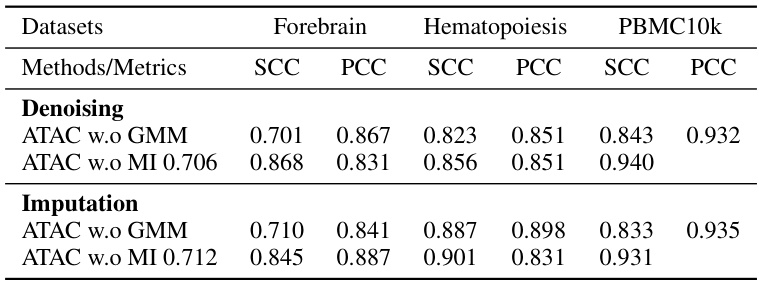
This table presents the results of denoising and imputation experiments performed using ATAC-Diff and other baseline methods on three scATAC-seq datasets (Forebrain, Hematopoiesis, and PBMC10k). The performance is measured using Spearman Correlation Coefficient (SCC) and Pearson Correlation Coefficient (PCC). Two ablation studies are shown, one without the Gaussian Mixture Model (GMM) and another without the mutual information (MI) component, highlighting the contribution of these components to the overall performance. Higher scores indicate better performance in both denoising (reducing noise) and imputation (filling in missing values) tasks.
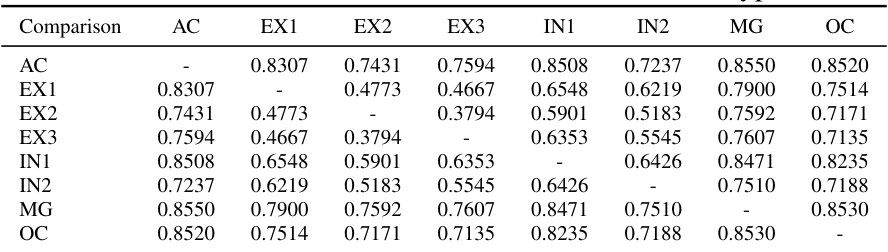
This table shows the Euclidean distances between the averaged latent embeddings of different cell types. The values represent the similarity between cell types, with lower values indicating higher similarity. This helps visualize how well the ATAC-Diff model separates different cell types in its latent space.
Full paper#