↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Integrating multi-omics single-cell data (like scRNA-seq and scATAC-seq) is crucial for understanding complex biological processes. However, obtaining large, labeled datasets for both modalities is often expensive and time-consuming. This creates a significant hurdle for researchers who need to analyze these datasets effectively. Existing knowledge transfer methods often require abundant labeled data, limiting their applicability in real-world scenarios.
The paper introduces DANCE, a semi-supervised knowledge transfer framework to address these limitations. DANCE uses optimal transport to generate pseudo-labels for scarce labeled scRNA-seq data and a divide-and-conquer strategy for scATAC-seq data. It further employs consistency regularization and a cross-omic multi-sample Mixup technique to reduce cell heterogeneity. Extensive experiments demonstrate DANCE’s superiority over existing methods, offering a more practical and efficient approach to multi-omic single-cell data integration.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in single-cell multi-omics because it tackles the problem of knowledge transfer under label scarcity, a common and expensive challenge in the field. It proposes a novel, semi-supervised method (DANCE) demonstrating superior performance over existing approaches. This opens new avenues for more efficient and cost-effective analyses, accelerating biological discovery.
Visual Insights#
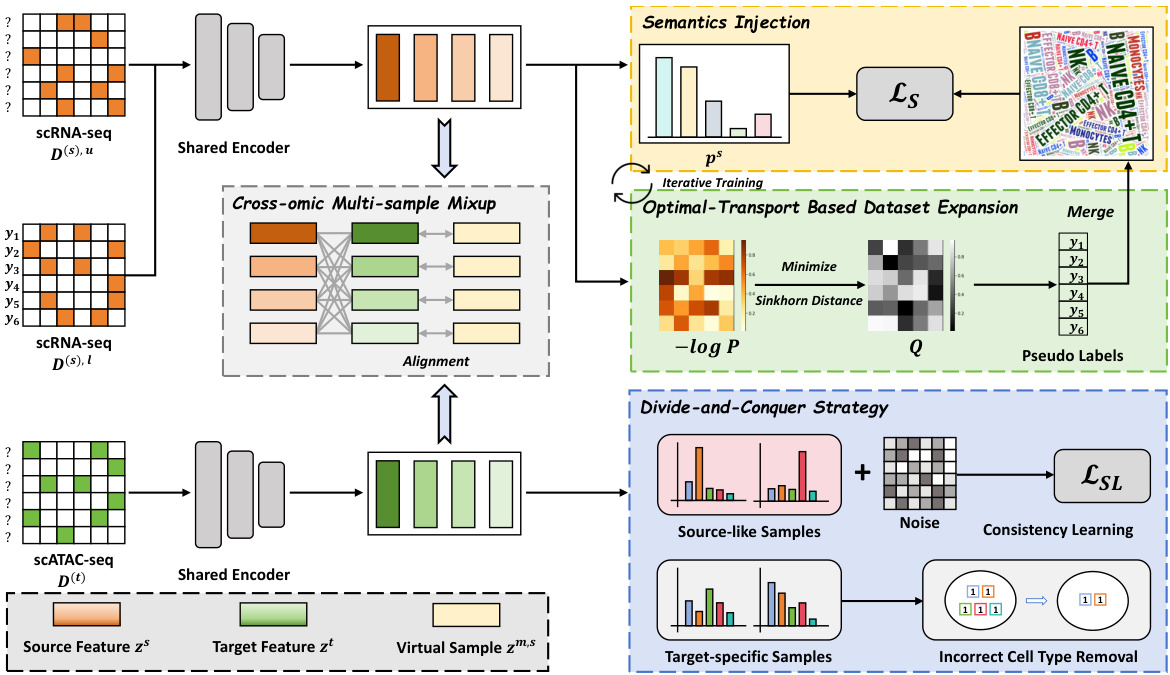
This figure provides a detailed overview of the DANCE framework. It illustrates the key steps involved in the semi-supervised knowledge transfer process, including semantics injection, dataset expansion using optimal transport, consistency regularization for source-like samples, ambiguous set learning for target-specific samples, and cross-omic multi-sample Mixup to reduce heterogeneity. Each step is visually represented with corresponding data and process components.
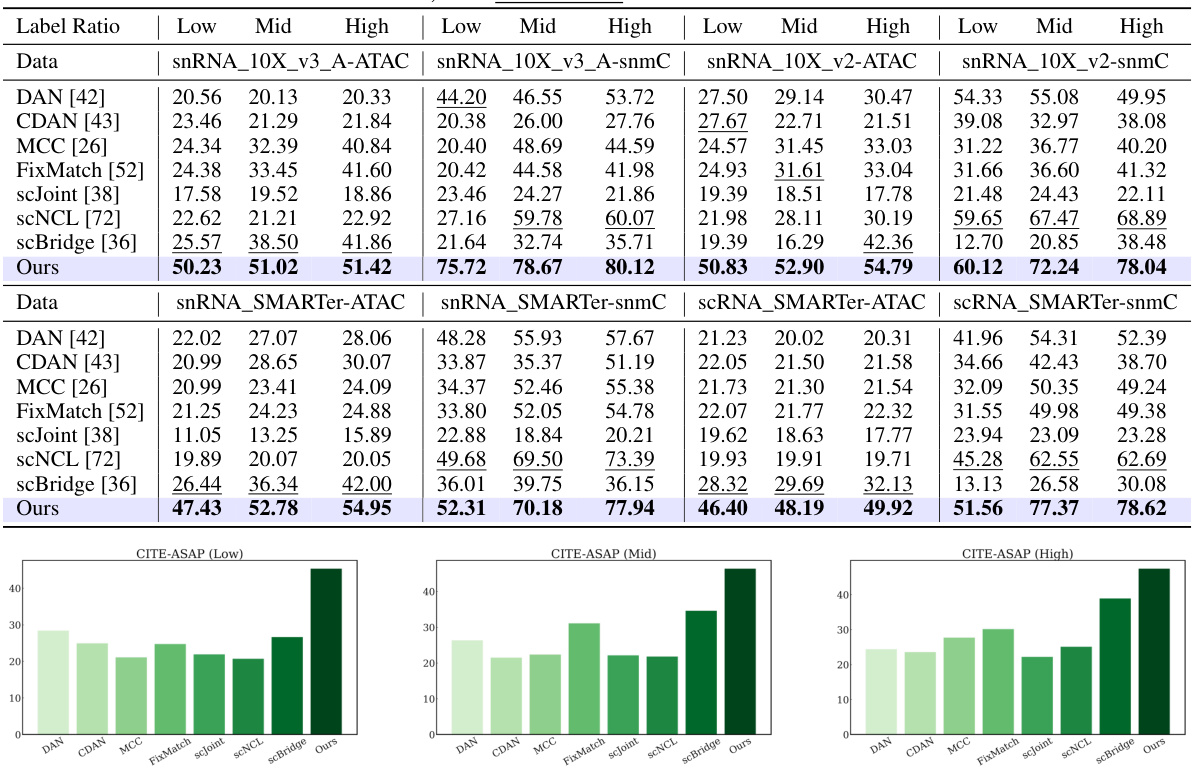
This table presents a quantitative comparison of the proposed DANCE method against several state-of-the-art approaches for knowledge transfer across multi-omic single-cell data. The comparison is performed under different label ratio conditions (Low, Mid, High) across four benchmark datasets. The results are expressed as percentages, with bold numbers highlighting the best performance and underlined numbers indicating the second-best performance for each dataset and label ratio.
In-depth insights#
Semi-Supervised Xfer#
Semi-supervised knowledge transfer tackles the challenge of leveraging limited labeled data alongside abundant unlabeled data for effective model training. It is particularly valuable in scenarios with high annotation costs, such as multi-omic single-cell data analysis where obtaining complete labels is expensive and time-consuming. This approach bridges the gap between fully supervised methods, which rely heavily on labeled data, and unsupervised methods, which lack the guidance of labels. The core idea is to use the labeled data to learn a robust representation and then transfer that knowledge to the unlabeled data, enabling the model to learn from both sources simultaneously. The success of semi-supervised transfer hinges on effective techniques to handle the heterogeneity present in the data and to avoid propagating errors from the labeled data to the unlabeled data. Methods often incorporate strategies like pseudo-labeling, consistency regularization, and self-training to achieve optimal knowledge transfer while mitigating the risk of overfitting and mislabeling.
Dual Label Scarcity#
The concept of “Dual Label Scarcity” highlights a critical challenge in multi-omic single-cell data analysis where both scRNA-seq and scATAC-seq data suffer from limited labeled samples. This scarcity significantly hinders the ability to effectively transfer cell type annotations from one modality (typically scRNA-seq) to the other (scATAC-seq). Traditional approaches often rely on abundant labeled data, making them impractical for this scenario. The dual scarcity necessitates innovative strategies that leverage both labeled and unlabeled data effectively, perhaps employing semi-supervised learning, pseudo-labeling techniques, or other methods to address the data imbalance problem. The core problem lies in extracting meaningful cellular information from limited annotations, leading to reduced accuracy and potentially biased downstream analyses. Solutions may involve advanced representation learning to maximize information extraction from scarce labels while simultaneously mitigating the heterogeneity that often exists between different omic data types. Overcoming dual label scarcity is therefore crucial for unlocking the full potential of multi-omic single-cell analysis, enabling more comprehensive and robust biological insights.
OT-Based Expansion#
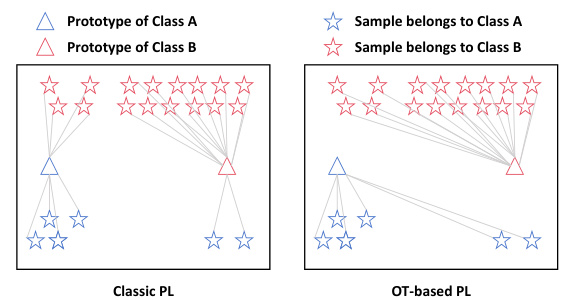
The Optimal Transport (OT)-based expansion method is a crucial part of the proposed framework. It cleverly addresses the challenge of label scarcity in single-cell RNA sequencing (scRNA-seq) data by generating pseudo-labels for unlabeled samples. This is achieved through the use of an optimal transport algorithm, which maps the low dimensional embedding space of the scRNA-seq data to a higher-dimensional space where cell type discrimination becomes more robust. This strategy is particularly useful in situations where obtaining high quantities of labeled data is expensive and time-consuming. Instead of simple sample-level prediction in traditional pseudo-labeling, OT incorporates a global perspective based on a cost function, minimizing the overall cost of transferring probabilities and generating a more accurate distribution of pseudo-labels. This approach demonstrably improves overall prediction performance and reduces bias. The integration of OT in this method is a significant advancement in semi-supervised knowledge transfer for single-cell multi-omic analysis.
Divide & Conquer#
A ‘Divide & Conquer’ strategy, in the context of multi-omic single-cell data analysis, likely involves partitioning the data into more manageable subsets to address inherent heterogeneity and label scarcity. This could entail separating scATAC-seq data into ‘source-like’ and ’target-specific’ groups, based on their similarity to the labeled scRNA-seq data. The source-like data, resembling the labeled data, can leverage techniques like consistency regularization, forcing consistent predictions across augmented samples. Target-specific data, distinct from the labeled source, would require a different approach, potentially involving iterative label refinement or the selection of ambiguous labels, progressively eliminating incorrect ones. This strategy acknowledges the differences in data characteristics and enables tailored processing for each subset, leading to a more robust and accurate cell type assignment.
Mixup Alignment#
Mixup Alignment, in the context of multi-omic single-cell data integration, presents a novel approach to address data heterogeneity. It leverages the concept of Mixup, a data augmentation technique, to create synthetic data points by interpolating features from different cells. This process helps to bridge the gap between scRNA-seq and scATAC-seq data, which often exhibit different feature distributions. The key insight is that by mixing features, the algorithm learns a more robust representation that captures common biological signals across modalities, reducing the impact of inherent technical biases and noise. The alignment aspect emphasizes generating these synthetic data points in a way that aligns the feature representations of the two different datasets, enhancing the ability to identify common cell types and ultimately improving the accuracy of cell type annotation transfer. This approach is particularly beneficial in semi-supervised settings, where labeled data is scarce, allowing the model to learn more effectively from the limited labeled information. While computationally more expensive than direct alignment methods, the potential improvement in accuracy and robustness makes Mixup Alignment a promising technique in this critical domain.
More visual insights#
More on figures

This figure provides a visual overview of the DANCE framework. It shows the different steps involved in the process: injecting semantics from labeled scRNA-seq data, expanding the dataset using optimal transport-based pseudo-labeling, employing consistency regularization and ambiguous set learning for scATAC-seq data (divided into source-like and target-specific samples), and finally using cross-omic multi-sample Mixup to address cell heterogeneity. Each step is represented visually, making it easier to understand the overall workflow of the proposed method.
This figure presents a flowchart illustrating the DANCE framework. It shows the process of injecting semantics from labeled scRNA-seq data, expanding the dataset using optimal transport, and handling scATAC-seq data through a divide-and-conquer strategy (source-like vs. target-specific samples). Consistency regularization and ambiguous label learning are applied to the respective sample types, and cross-omic multi-sample Mixup is used to reduce cell heterogeneity.
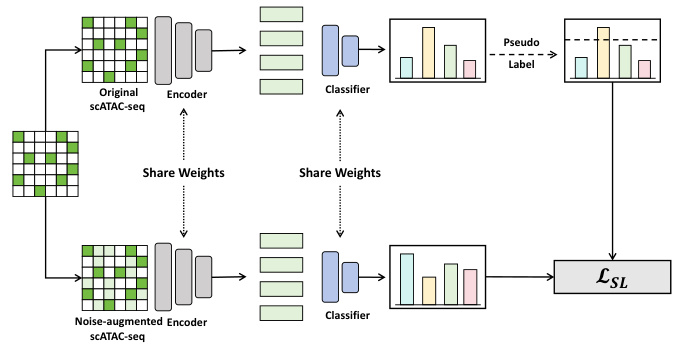
This figure illustrates the DANCE framework. It starts with labeled scRNA-seq data to inject semantics. It uses optimal transport for pseudo-labeling of unlabeled scRNA-seq data and expands the dataset. The scATAC-seq data is divided into source-like and target-specific data. Consistency regularization is applied to source-like samples while target-specific samples undergo ambiguous set learning. Finally, cross-omic multi-sample mixup is used to reduce cell heterogeneity, ultimately improving cell type transfer from scRNA-seq to scATAC-seq data.

This figure provides a visual overview of the DANCE framework. It illustrates the different stages of the process, starting from semantics injection and dataset expansion using optimal transport, to the divide-and-conquer strategy for handling source-like and target-specific scATAC-seq data, and finally, the application of cross-omic multi-sample Mixup to reduce cell heterogeneity. Each stage is represented by a distinct block in the diagram, showing the flow of data and the key operations involved.

This figure provides a visual overview of the DANCE framework. It illustrates the different steps involved in the process, starting with semantics injection and dataset expansion using labeled scRNA-seq data and optimal transport. The framework then uses a divide-and-conquer strategy for scATAC-seq data, employing consistency regularization and ambiguous set learning for different subsets of the data. Finally, it utilizes cross-omic multi-sample Mixup to reduce cell heterogeneity.
This figure provides a visual overview of the DANCE framework’s workflow. It begins by using labeled scRNA-seq data to inject semantic knowledge into the model. It addresses the challenge of label scarcity by expanding the scRNA-seq dataset using optimal transport (OT) to generate pseudo-labels. The scATAC-seq data is then divided into source-like and target-specific subsets, and different methods are applied for each subset. The source-like samples utilize consistency regularization with random perturbations, while the target-specific samples use an iterative approach to eliminate incorrect labels. Finally, to mitigate the cell heterogeneity between the two datasets, a cross-omic multi-sample mixup technique is employed. This creates virtual samples that bridge the gap between the data modalities, leading to a more robust and accurate cell type transfer.
This figure provides a visual overview of the DANCE framework. It illustrates the main steps involved in the semi-supervised knowledge transfer process. It starts with labeled scRNA-seq data, uses optimal transport for pseudo-labeling, and then employs consistency regularization and ambiguous set learning for the scATAC-seq data. Finally, it utilizes cross-omic multi-sample Mixup to reduce heterogeneity.

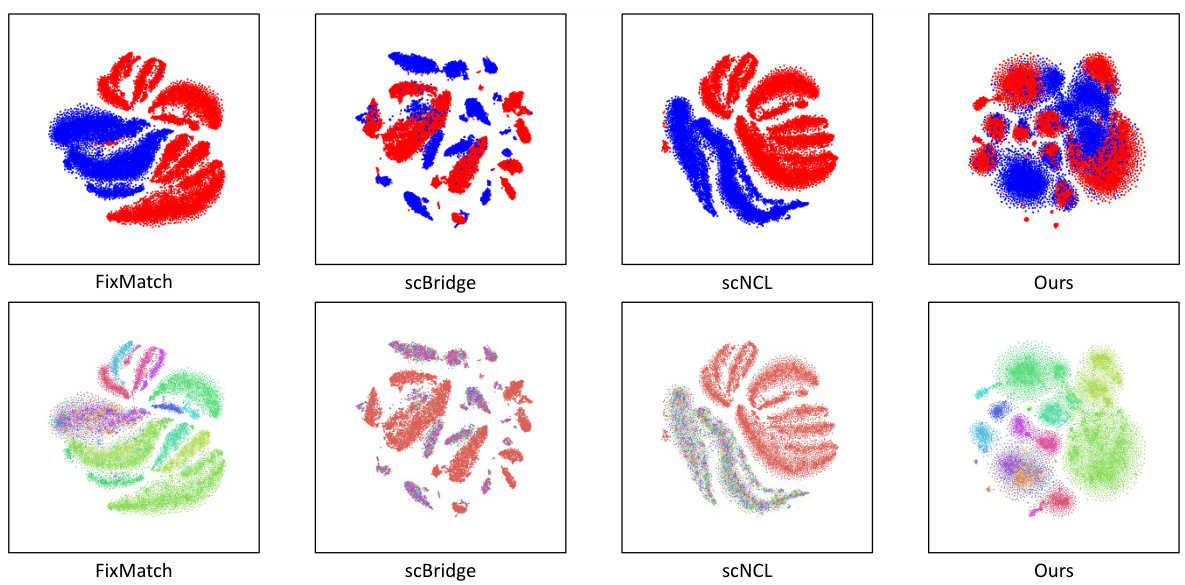
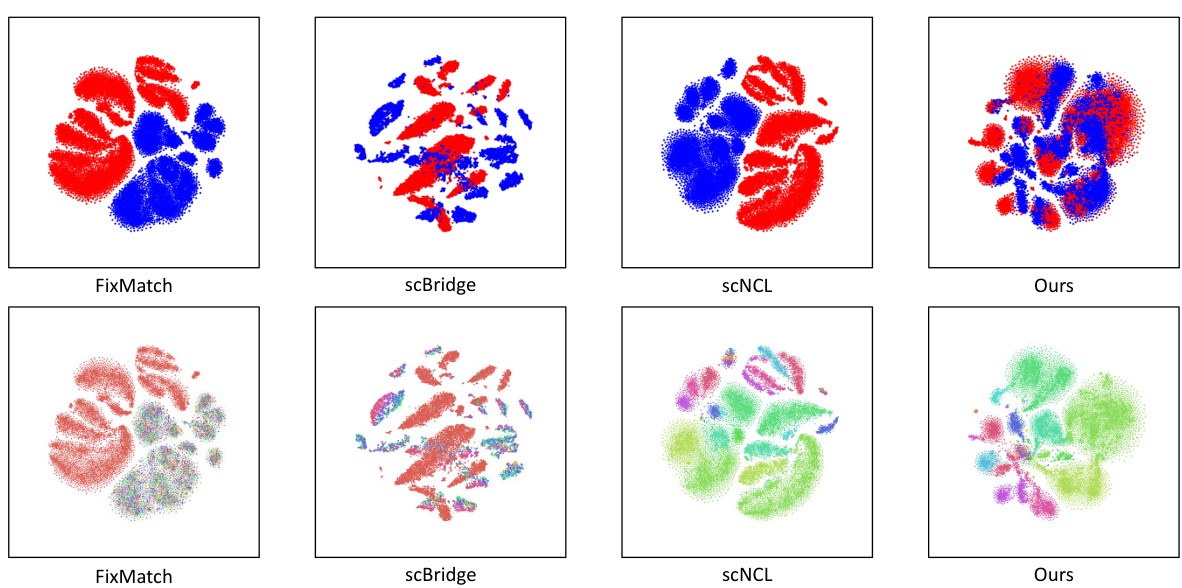
This figure shows the results of several experiments to evaluate the performance of the proposed method. (a) shows the sensitivity analysis of the hyperparameters k and λ. (b) shows the t-SNE visualization of the two modalities with a high label ratio. (c) shows the A-distance comparison with different label ratios. The results indicate that the proposed method is robust and effective in reducing cell heterogeneity.
More on tables

This table presents the results of ablation studies performed on the proposed DANCE model. By removing different components of the model (Optimal Transport, source-like sample learning, target-specific sample learning, and cross-omic multi-sample mixup), the impact of each component on the overall performance is evaluated across different datasets and label ratios (low, mid, high). The ‘Full Model’ row shows the performance of the complete DANCE model, serving as a benchmark.
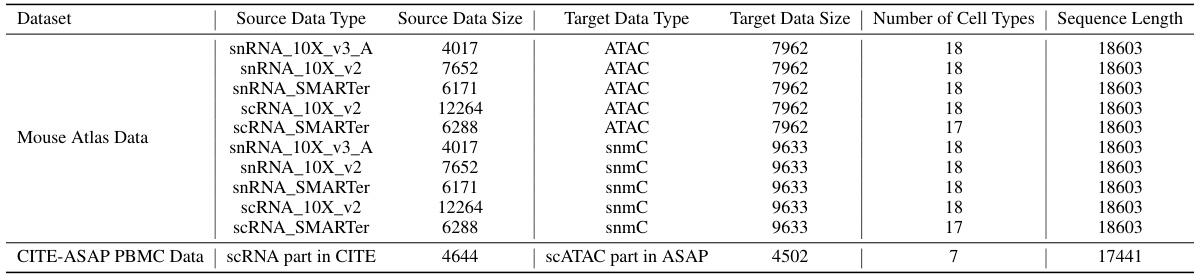
This table shows the details of the datasets used in the experiments, including the source data type, source data size, target data type, target data size, the number of cell types, and the sequence length for each dataset. The datasets include various combinations of scRNA-seq and scATAC-seq or scRNA-seq and snmC data, with varying numbers of cells and sequence lengths. This information is crucial for understanding the scope and reproducibility of the experimental results.

This table presents the results of a sensitivity analysis performed to evaluate the impact of the hyperparameter σ on the performance of the proposed method. The analysis was conducted on two different datasets, CITE-ASAP and snRNA_10X_v3_A-ATAC, each with different values of σ (5, 10, 15, 20). The results indicate that the model’s performance is relatively stable across different values of σ, showing its robustness to changes in this hyperparameter.

This table shows the results of a sensitivity analysis on the hyperparameter τ. The analysis was performed on the CITE-ASAP and snRNA_10X_v3_A-ATAC datasets. Different values of τ (0.8, 0.85, 0.9, 0.95) were tested and the corresponding accuracy is reported. The purpose of this analysis was to determine the sensitivity of the model’s performance to changes in τ, and to identify an optimal value for this parameter.

This table presents the results of an ablation study analyzing the sensitivity of the proposed DANCE model’s performance to variations in the threshold parameter μ. The study was conducted on two datasets: CITE-ASAP and snRNA_10X_v3_A-ATAC. The table shows that the model’s performance is relatively stable across different values of μ, with peak performance observed around 1e-3. This suggests that DANCE is not overly sensitive to the choice of μ, enhancing its robustness and practical applicability.

This table presents the ablation study results focusing on the impact of the Optimal Transport (OT)-based strategy when applied to target scATAC-seq data. It compares the performance of the model with and without the OT strategy across different label ratios (Low, Mid, High), demonstrating the effectiveness of the OT-based approach for improving the accuracy of cell type prediction in scATAC-seq data.
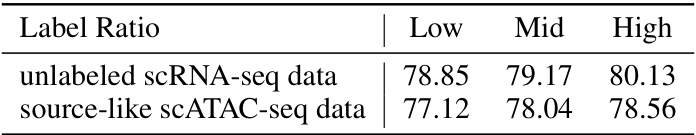
This table compares the accuracy of pseudo-labeling for unlabeled scRNA-seq data and source-like scATAC-seq data across different label ratios (Low, Mid, High). It demonstrates that the pseudo-labeling accuracies are quite similar between the two types of data, supporting the effectiveness of the proposed method’s approach to handling label scarcity in both datasets.

This table presents a comparison of the performance of several methods when transferring cell types from scATAC-seq data to scRNA-seq data. The results show the accuracy of each method under different label ratios (Low, Mid, High). The ‘Ours’ method consistently outperforms all the other methods.

This table compares the memory cost and training time per epoch for four different methods: scJoint, scBridge, scNCL, and the proposed method (Ours). The accuracy achieved by each method is also shown. The proposed method demonstrates comparable efficiency to the other methods while achieving significantly higher accuracy.
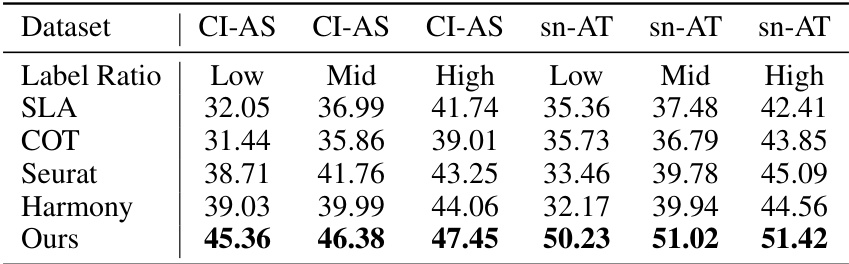
This table presents the ablation study results for the proposed DANCE model. It shows the performance of DANCE when different components are removed to assess their individual contributions. The results are presented for different label ratios (low, mid, and high) on multiple datasets. The table helps determine the importance of each module within the DANCE framework.
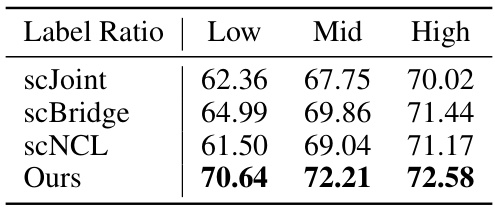
This table presents the performance comparison of different methods (scJoint, scBridge, scNCL, and the proposed DANCE method) on the full MouseAtlas dataset. The results are shown for different label ratios (Low, Mid, High), indicating the accuracy of cell type prediction under varying amounts of labeled data. The table highlights the performance gains achieved by DANCE method compared to existing state-of-the-art approaches across different label ratio settings.

This table presents the quantitative results of the proposed DANCE framework and several state-of-the-art methods on real-world scenarios. It shows the performance (accuracy in %) achieved when transferring cell type knowledge from labeled and unlabeled scRNA-seq data to scATAC-seq data. The results demonstrate DANCE’s superior performance in realistic settings where only a limited amount of labeled data is available.
Full paper#