↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Drug discovery heavily relies on virtual screening to identify promising drug candidates from vast molecular libraries. Existing ligand-based methods often struggle with limited labeled data and noisy information, hindering their ability to effectively explore the vast chemical space. Structure-based methods are also limited by the availability of protein target structures. This creates a significant challenge in finding effective and efficient approaches for drug discovery.
This paper introduces S-MolSearch, a novel framework that addresses these limitations. It uses semi-supervised contrastive learning and incorporates 3D molecular information to efficiently learn from both labeled and unlabeled data. The use of inverse optimal transport ensures adaptive utilization of unlabeled data. S-MolSearch demonstrates superior performance over existing methods on widely used benchmarks, showcasing its efficacy in bioactive molecule search and the potential for significant advancement in drug discovery.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in drug discovery and cheminformatics. It presents S-MolSearch, a novel and highly effective framework for bioactive molecule search, significantly advancing virtual screening techniques. The semi-supervised contrastive learning approach, combined with inverse optimal transport, offers superior performance over existing methods and opens new avenues for leveraging both labeled and unlabeled data effectively in virtual screening. This work’s impact is heightened by its focus on 3D molecular information and adaptability to diverse data. It is particularly relevant given the limitations of current structure-based and ligand-based approaches.
Visual Insights#
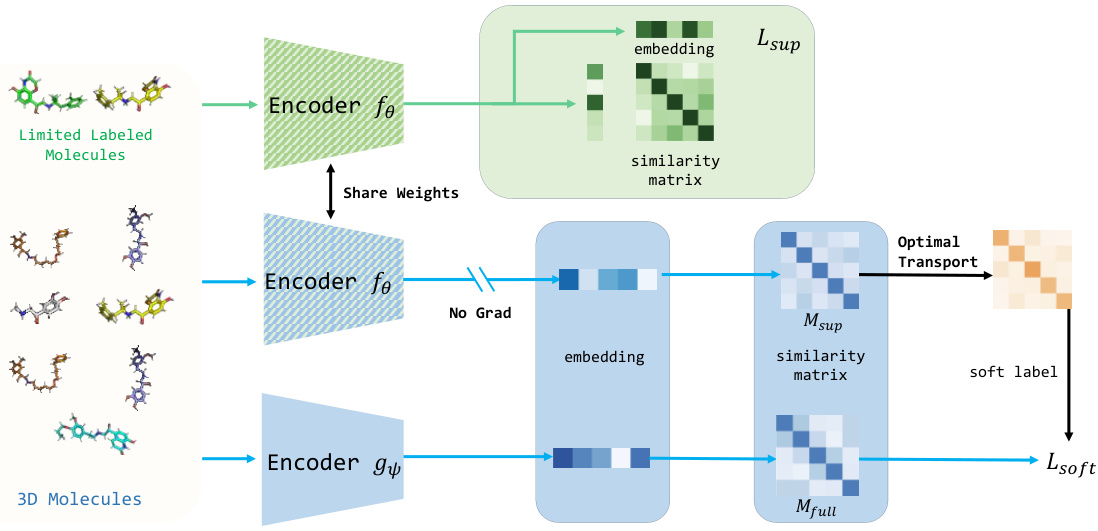
This figure illustrates the architecture of the S-MolSearch framework. It shows two encoders, fe and gy. Encoder fe processes limited labeled molecules using contrastive learning to generate a similarity matrix and embeddings. Encoder gy processes the full dataset (labeled and unlabeled molecules). The embeddings from fe, without gradient updates, are used to compute a similarity matrix. Optimal transport is then used to transfer information from fe to gy, generating soft labels for gy which are used in training along with the full data similarity matrix. The framework combines semi-supervised contrastive learning with optimal transport to leverage both labeled and unlabeled data for more effective molecule search.

This table presents the performance comparison of S-MolSearch against various baselines (ROCS, Phase Shape, LIGSIFT, SHAFTS, Glide-SP, Vina, Paafnucy, OnionNet, Planet, DrugCLIP) on the DUD-E benchmark dataset. The evaluation is performed in a zero-shot setting, meaning the models are not fine-tuned on the DUD-E dataset. Performance is measured using AUROC (Area Under the Receiver Operating Characteristic curve), BEDROC (Boltzmann-Enhanced Discrimination of Receiver Operating Characteristic curve), and EF (Enrichment Factor) at different thresholds (0.5%, 1%, and 5%). S-MolSearch, with two variations (trained with a 0.4 similarity threshold and a 0.9 similarity threshold), demonstrates superior performance across all metrics compared to the other methods.
In-depth insights#
3D Mol Search#
A hypothetical “3D Mol Search” system, as described in the context of the provided research paper, would likely leverage 3D structural information of molecules alongside affinity data, employing semi-supervised contrastive learning techniques. The method would likely involve two encoders: one trained on labeled data to capture affinity-based relationships, and another trained on both labeled and unlabeled data, utilizing inverse optimal transport to efficiently integrate the information from both sources. The core innovation would likely be the fusion of 3D-structural and affinity information in a semi-supervised framework that facilitates improved representation learning for better virtual screening performance. This approach addresses limitations of traditional methods which either focus exclusively on 2D representations or exclusively on structure-based methods, offering a more comprehensive and robust solution to virtual screening, particularly helpful when labeled data is limited and noisy. The system’s effectiveness would depend heavily on the performance of the encoder networks and the effectiveness of the inverse optimal transport integration.
Semi-Supervised IOT#
A semi-supervised approach using Inverse Optimal Transport (IOT) for molecular property prediction offers a powerful strategy to leverage both labeled and unlabeled data. The scarcity of labeled data in many cheminformatics tasks necessitates this approach. IOT, by learning a cost function that aligns probability distributions of labeled and unlabeled data, elegantly addresses the challenge of learning from limited supervision. This method can effectively extract meaningful representations from unlabeled data, improving model generalization and predictive accuracy. Incorporating 3D structural information, as is common in molecular applications, makes the framework even more powerful, particularly when combined with contrastive learning methods for distinguishing active and inactive molecules. The benefits include enhanced robustness to noisy or inconsistent data, which is often a problem with experimental affinity measurements, and the ability to explore a larger chemical space without relying solely on expensive and time-consuming experiments. Ultimately, a semi-supervised IOT framework provides a significant advance in the efficiency and effectiveness of virtual screening and molecular property prediction.
Benchmark Results#
A dedicated section on benchmark results would be crucial for evaluating the performance of S-MolSearch. It should clearly present results across multiple widely-accepted datasets, such as DUD-E and LIT-PCBA, comparing S-MolSearch against a range of state-of-the-art baselines (both ligand-based and structure-based methods). Key metrics like AUROC, BEDROC, and EF should be reported, not just as single numbers but with error bars or confidence intervals to indicate statistical significance and reliability of the findings. Furthermore, a discussion of these results is essential, highlighting S-MolSearch’s strengths (e.g., superior performance in certain scenarios or datasets) and weaknesses (e.g., lower performance in others), and offering plausible explanations. The analysis should ideally cover different training parameters or data splits to show how sensitive the model is to these choices. A compelling benchmark section would firmly establish S-MolSearch’s position in the field, providing a strong foundation for future research and applications.
Ablation Study#
An ablation study systematically evaluates the contribution of individual components within a machine learning model. In the context of the S-MolSearch paper, this would involve removing or disabling specific parts of the framework (e.g., the semi-supervised component, the inverse optimal transport mechanism, or specific regularization techniques) to assess their impact on the overall performance. The goal is to determine which parts are essential for achieving state-of-the-art results and which ones might be redundant or even detrimental. By isolating the effects of each component, the ablation study provides valuable insights into the model’s architecture and identifies potential areas for improvement or simplification. The results of this process might reveal unexpected interactions between components, or suggest alternative design choices that yield similar performance with reduced computational complexity. Such analysis is critical for ensuring the robustness and reproducibility of the findings, while also informing future model development.
Future Works#
The authors suggest several promising avenues for future research. Improving the interpretability of S-MolSearch is a key area; while the model achieves state-of-the-art performance, understanding its decision-making process remains opaque. This could involve developing techniques to visualize the learned representations, potentially linking them to specific molecular features or interactions. Incorporating more comprehensive datasets is another vital direction. While ChEMBL provides rich information, including other data modalities like protein-protein interaction networks, or integrating data from multiple sources, could lead to more robust and generalizable models. Exploring different contrastive learning strategies beyond the semi-supervised approach presented could yield further improvements in accuracy and efficiency. Finally, extending S-MolSearch to other virtual screening tasks beyond ligand-based identification presents a significant opportunity. This might entail adapting the model to accommodate structure-based methods or to address other challenging aspects of drug discovery.
More visual insights#
More on figures
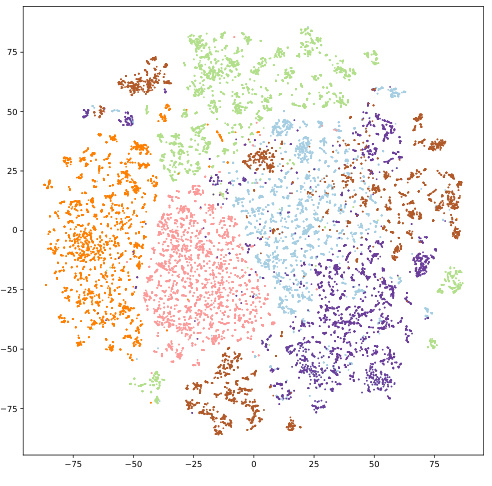
This figure compares the t-distributed Stochastic Neighbor Embedding (t-SNE) visualizations of molecular representations generated by two different approaches: (a) using a pretrained checkpoint (Uni-Mol), and (b) using the S-MolSearch model. Different colors represent molecules that bind to different protein targets. The visualization aims to show how well the different methods separate molecules according to their target protein. The S-MolSearch model shows clearer separation between different targets and tighter clustering within each target, suggesting that it learns more informative representations that better capture the relationships between molecular structure and activity.

This figure compares the t-SNE visualizations of molecular representations learned by the S-MolSearch model and a pretrained checkpoint. The visualizations show how well the model separates molecules based on their protein targets. Different colors represent different protein targets. The S-MolSearch model shows a better separation of the molecules indicating that it better captures the relationships between molecules and their protein targets.

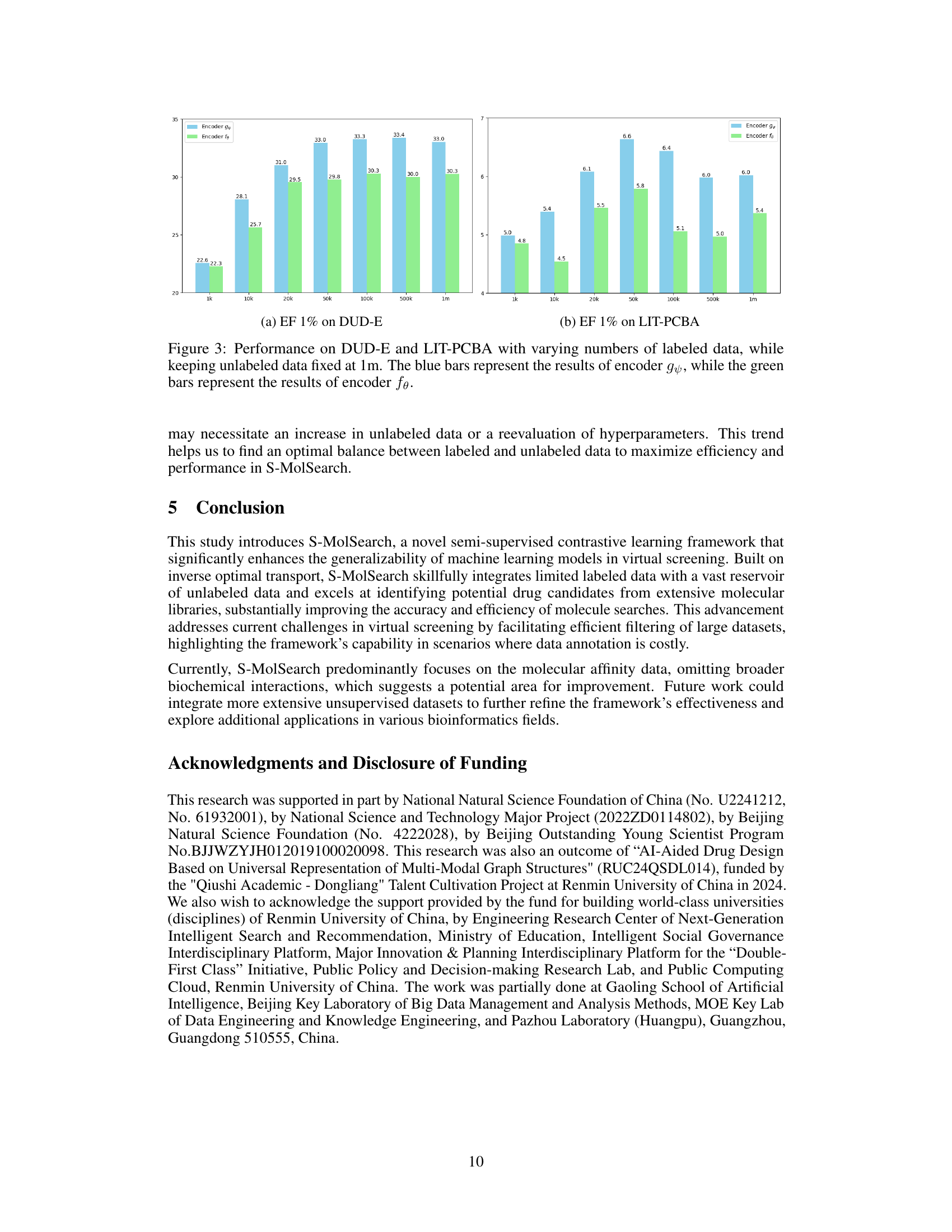
This figure shows the performance of both encoder fθ (trained only on labeled data) and encoder gψ (trained on both labeled and unlabeled data) on the DUD-E and LIT-PCBA datasets. The x-axis represents the amount of labeled data used for training, ranging from 1k to 1m. The y-axis shows the EF1% (Enrichment Factor at 1% of the database screened), a metric evaluating the effectiveness of virtual screening. The results clearly demonstrate that using unlabeled data significantly improves performance (blue bars consistently outperform green bars) and that there is an optimal amount of labeled data for best performance. Beyond a certain amount, increasing labeled data doesn’t improve results.
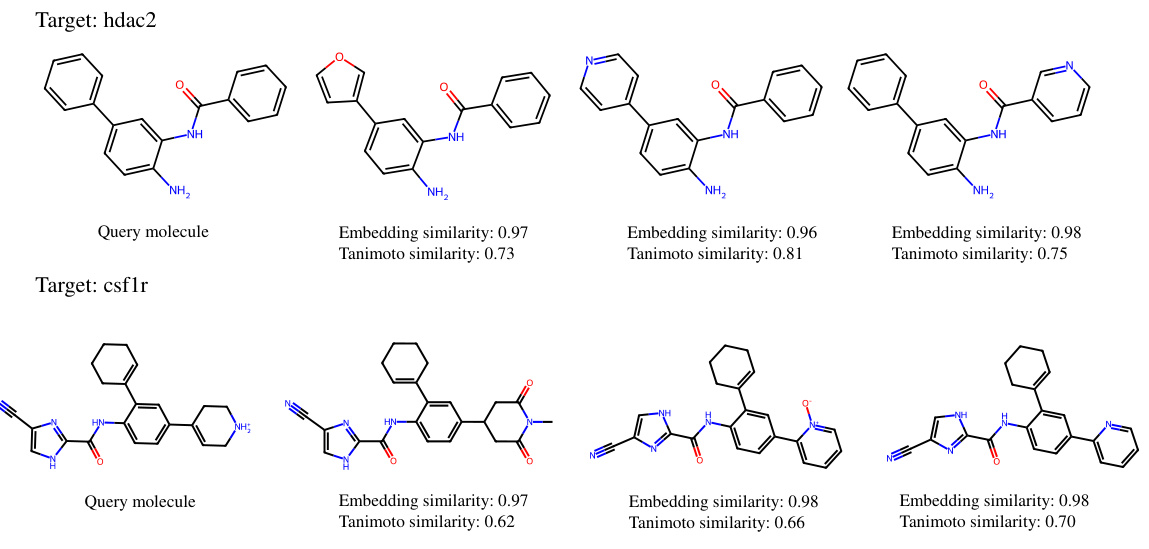
This figure shows examples of similar molecules retrieved by S-MolSearch for two target proteins (hdac2 and csflr) from the DUD-E dataset. For each target, the query molecule is shown alongside its top-ranked similar molecules. The embedding similarity (calculated by S-MolSearch) and Tanimoto similarity (a traditional measure of molecular similarity) are provided for each pair, demonstrating the correlation between the two similarity metrics.
More on tables
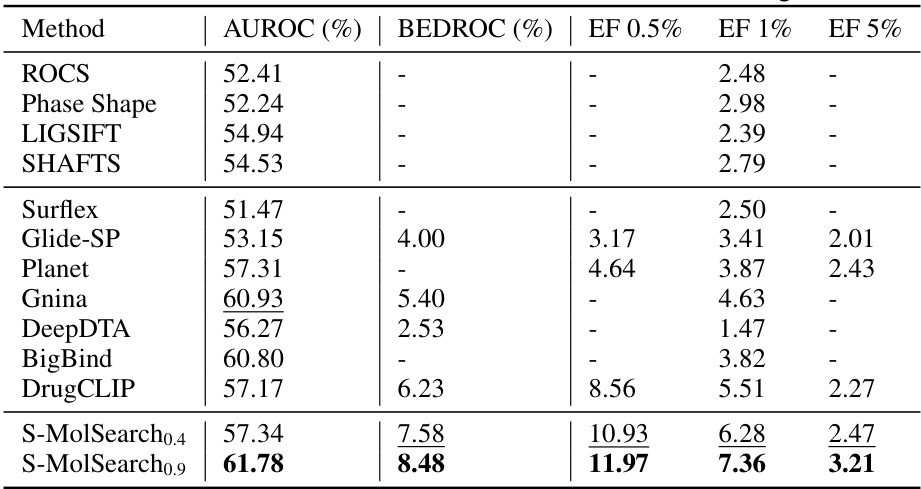
This table presents the performance of various virtual screening methods on the LIT-PCBA benchmark dataset in a zero-shot setting (i.e., without any fine-tuning on the benchmark dataset). The metrics used for evaluation are AUROC (Area Under the Receiver Operating Characteristic curve), BEDROC (Boltzmann-Enhanced Discrimination of Receiver Operating Characteristic curve), and Enrichment Factor at 0.5%, 1%, and 5% (EF0.5%, EF1%, EF5%). Higher values for these metrics indicate better performance. The table compares S-MolSearch’s performance (with two different similarity thresholds: 0.4 and 0.9) to several established ligand-based (LBVS) and structure-based (SBVS) virtual screening methods. This allows for a direct comparison and assessment of S-MolSearch’s effectiveness relative to existing state-of-the-art techniques. The zero-shot setting is crucial as it reflects the practical scenario of using a pre-trained model without dataset-specific adaptation.

This table presents the results of ablation studies conducted on the S-MolSearch model using the DUD-E and LIT-PCBA benchmark datasets. The study systematically removes components of the model (soft label, regularizer, and pretrain) to evaluate their individual contribution to the overall performance. The results are presented as enrichment factors (EF) at 0.5%, 1%, and 5% thresholds for both datasets, showing the impact of each removed component on the model’s ability to identify active molecules.

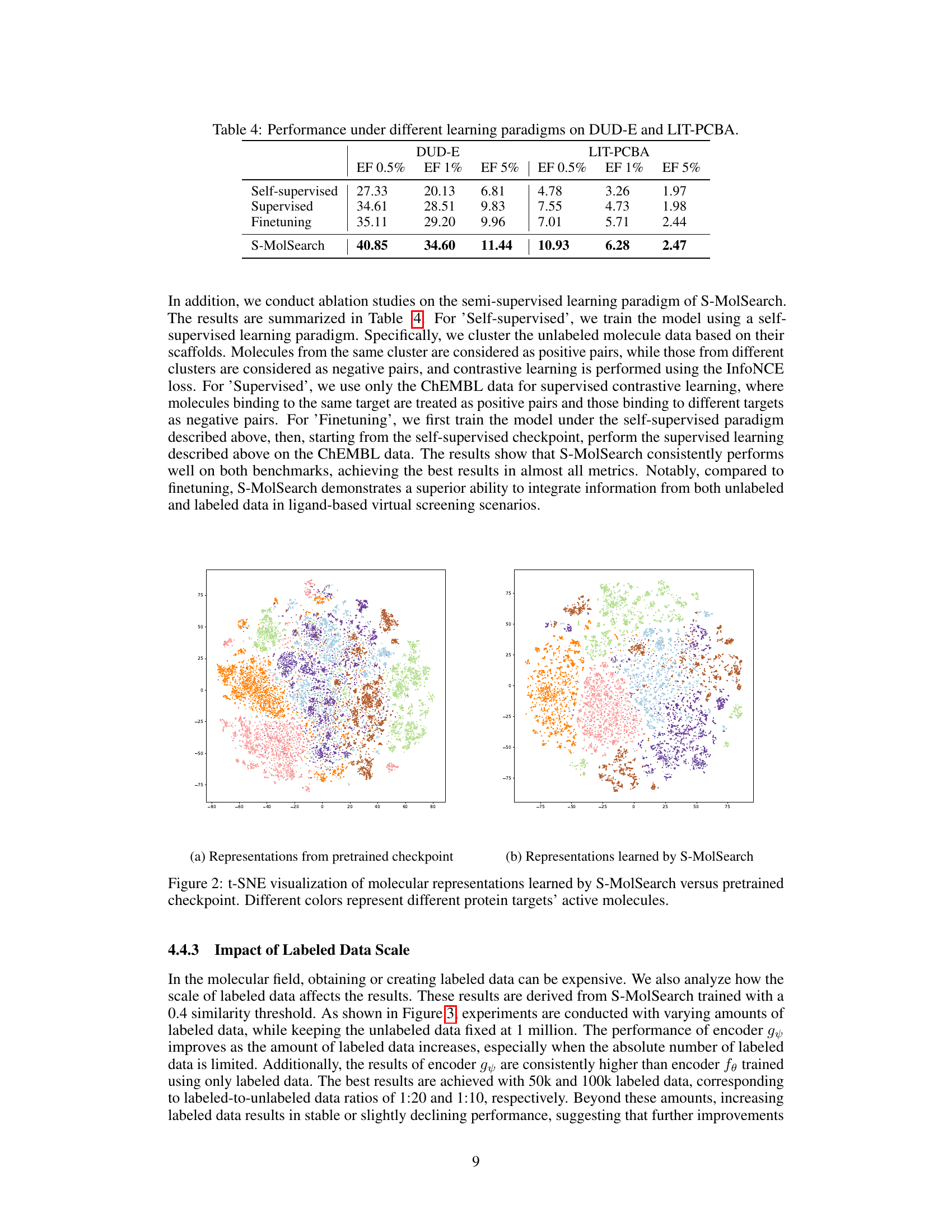
This table compares the performance of different learning approaches (self-supervised, supervised, finetuning, and S-MolSearch) on two virtual screening benchmarks, DUD-E and LIT-PCBA. The metrics used are enrichment factors (EF) at different thresholds (0.5%, 1%, and 5%). The results demonstrate the effectiveness of S-MolSearch compared to other methods in integrating both labeled and unlabeled data for improved virtual screening performance.
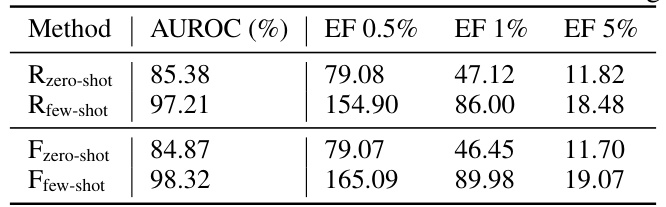
This table presents the performance of S-MolSearch on the DUD-E benchmark in two different few-shot learning settings. It compares the performance of the zero-shot setting (where the model is not trained on any data from the benchmark) against two few-shot settings (where a small subset of the benchmark’s data is used for training). The results are shown in terms of AUROC, EF 0.5%, EF 1%, and EF 5%. Two different few-shot methodologies (R and F) are compared.
Full paper#