↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Identifying protein active sites is crucial for drug discovery and understanding enzyme mechanisms. Traditional methods are labor-intensive, while existing computational approaches struggle due to limited functional annotations, particularly precise per-residue data. This scarcity hinders the performance of protein language models (PLMs), which mainly rely on amino acid sequences.
To overcome these limitations, the researchers propose MMSite, a multi-modal framework that integrates both protein sequences and rich textual descriptions. MMSite employs a “First Align, Then Fuse” strategy, aligning the textual modality with the sequential modality using soft-label alignment and then identifying active sites via multi-modal fusion. The use of a MACross module effectively handles the multi-attribute nature of textual descriptions. Experimental results demonstrate MMSite’s superior performance compared to existing methods, showcasing the potential of multi-modal learning in this critical area of biological research. The dataset and code implementation are publicly available.
Key Takeaways#
Why does it matter?#
This paper is significant because it introduces a novel multi-modal framework, MMSite, for identifying protein active sites, a crucial task in drug discovery and life sciences. MMSite leverages both protein sequences and rich textual descriptions, improving upon existing methods and achieving state-of-the-art performance. This opens new avenues for research in multi-modal protein analysis and improves the accuracy of active site prediction, impacting drug design and fundamental biological understanding.
Visual Insights#

This figure highlights the key difference between existing protein representation learning methods and the proposed MMSite framework. Existing methods primarily focus on sequence-level tasks like function prediction or text generation from sequences, often neglecting residue-level details. In contrast, MMSite directly tackles the identification of active sites at the residue level, utilizing both protein sequences and rich multi-attribute textual descriptions to achieve a more comprehensive and detailed understanding.
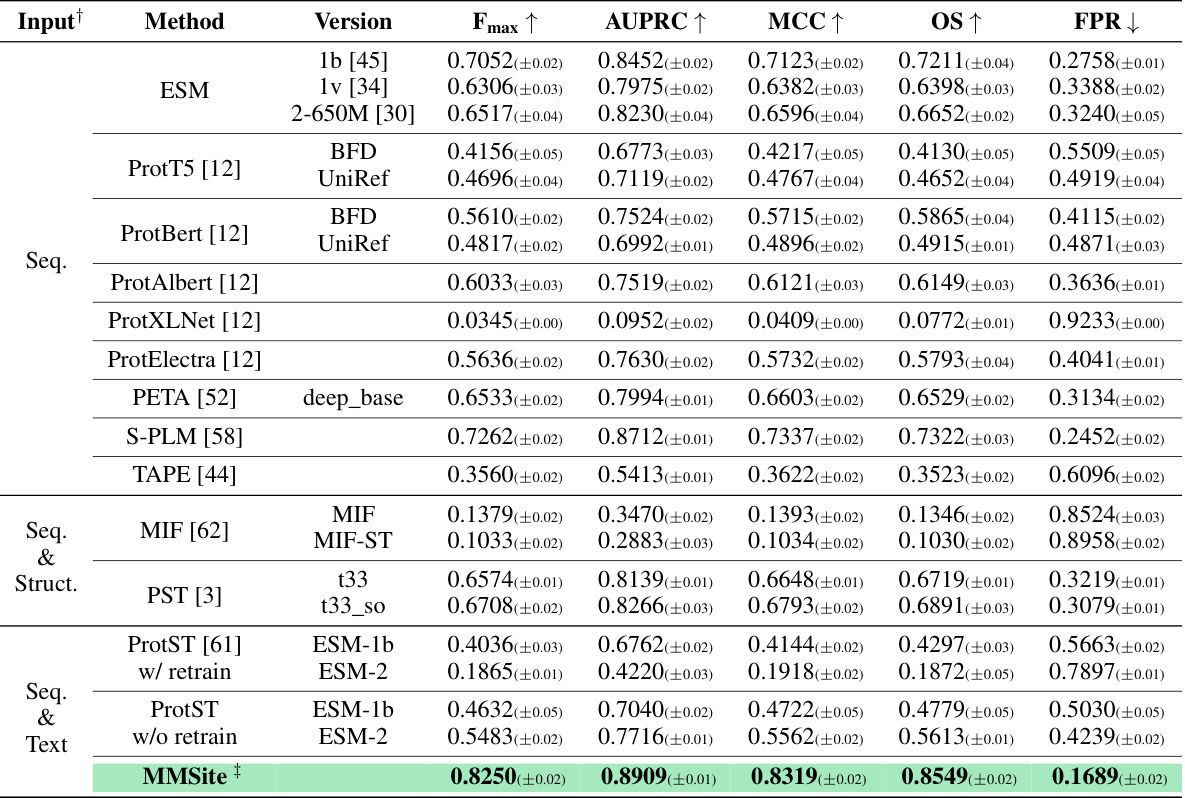
This table presents a comparison of the MMSite model’s performance against 21 other protein representation learning (PRL) models. The comparison is made using several metrics (Fmax, AUPRC, MCC, OS, FPR) to evaluate the accuracy of active site identification. The dataset used for the comparison is the ProTAD dataset with a clustering threshold of 10%. The table also indicates the input modality (sequence only, sequence and structure, sequence and text) used by each model. The results highlight MMSite’s superior performance in this specific task.
In-depth insights#
Multi-modal protein analysis#
Multi-modal protein analysis represents a significant advancement in the field of proteomics. By integrating diverse data modalities such as amino acid sequences, protein structures (3D or 2D), and associated textual information (functional annotations, experimental descriptions), researchers can gain a more holistic and comprehensive understanding of protein function and behavior. This approach offers enhanced predictive capabilities, surpassing the limitations of single-modality analysis. The integration of diverse data types enables more robust and accurate modeling of protein properties, interactions, and dynamics, potentially leading to improved drug discovery, enzyme engineering, and disease diagnostics. Challenges in multi-modal protein analysis include the development of effective methods for data fusion and integration, addressing the heterogeneity and variability of different data sources, and handling the scarcity of high-quality annotated data. Despite these challenges, the field is rapidly advancing, with the emergence of powerful deep learning techniques that leverage the synergistic information provided by multiple data types. The future of multi-modal protein analysis is promising, with the potential to revolutionize our understanding of proteins and their biological roles.
ProTAD dataset#
The ProTAD dataset represents a significant contribution to the field of protein active site prediction. Its construction involved compiling over 570,000 protein sequences paired with rich, multi-attribute textual descriptions. This pairing of sequence data with detailed textual annotations (17 distinct attributes) is crucial, addressing the scarcity of precise per-residue annotations that often limits the effectiveness of protein language models. The high-quality and multifaceted nature of the textual descriptions significantly enhances the dataset’s value for training multi-modal models, allowing these models to learn richer representations of proteins and their functional characteristics. The inclusion of attributes such as protein function, organism, and cautions enhances the biological relevance and applicability of the dataset. Furthermore, the careful data cleaning and rigorous quality control measures employed during the dataset’s creation ensures data reliability and minimize bias, further enhancing its value for reliable and robust model training. The dataset’s availability through a public GitHub repository promotes open science and facilitates wider adoption of advanced multi-modal active site prediction techniques.
MMSite framework#
The MMSite framework represents a novel multi-modal approach to protein active site identification. It leverages both protein sequence data (through protein language models) and rich multi-attribute textual descriptions (processed by biomedical language models) to achieve superior performance. A key innovation is the MACross module, which handles the multi-attribute nature of textual data effectively. The framework employs a two-stage process: first aligning the textual and sequential modalities using soft-label alignment, then fusing them for active site prediction. This approach tackles the data scarcity challenge common in per-residue protein annotations. The use of prompting and the alignment strategy are crucial for improving PLM performance in this context. The results demonstrate that MMSite outperforms existing methods, highlighting its effectiveness and potential for advancing life sciences and drug discovery.
Alignment & fusion#
Alignment and fusion are crucial steps in multi-modal learning, aiming to integrate information from different modalities effectively. Alignment seeks to establish correspondence between features from disparate sources, for example, aligning protein sequences with their textual descriptions. This might involve techniques like contrastive learning to learn shared representations or attention mechanisms to focus on relevant features in each modality. Fusion, on the other hand, combines the aligned representations to create a unified, holistic representation that is richer and more informative than either modality alone. This might involve simple concatenation, attention-based fusion or more sophisticated neural network architectures. The success of these methods heavily depends on the quality of the alignment: poorly aligned inputs will severely limit the effectiveness of the fusion process. Therefore, choosing appropriate alignment strategies tailored to the specific characteristics of the modalities is crucial. Finally, the choice of fusion technique should consider the desired trade-off between computational efficiency and the ability to capture complex interactions between modalities. A well-designed alignment and fusion scheme is key to unlocking the power of multi-modal data and achieving superior performance in downstream tasks like protein active site identification.
Future research#
Future research directions stemming from this work could explore enhancing the multi-modal framework’s capabilities. Improving the text generation agent’s accuracy would reduce reliance on existing annotations. Exploring alternative modalities, such as incorporating protein structure information or experimental data, could enrich the model and enhance predictions. The framework’s robustness to noisy or incomplete data needs further investigation. Addressing the computational cost, particularly for large-scale applications, would be important. Finally, applying MMSite to other biologically relevant tasks, such as predicting protein-protein interactions or drug-target binding, would showcase its wider applicability and contribute to our understanding of biological mechanisms.
More visual insights#
More on figures

This figure provides a detailed overview of the MMSite framework, a multi-modal approach for identifying active sites in proteins. It illustrates the three main stages of the framework: feature extraction, semantic alignment, and fusion and prediction. The figure also highlights the use of protein language models (PLMs), biomedical language models (BLMs), manual prompting, and a multi-attribute cross-attention module (MACross) to process protein sequences and multi-attribute textual descriptions. It showcases the ‘First Align, Then Fuse’ strategy used for aligning and fusing the dual modalities and explains how the model predicts active sites.
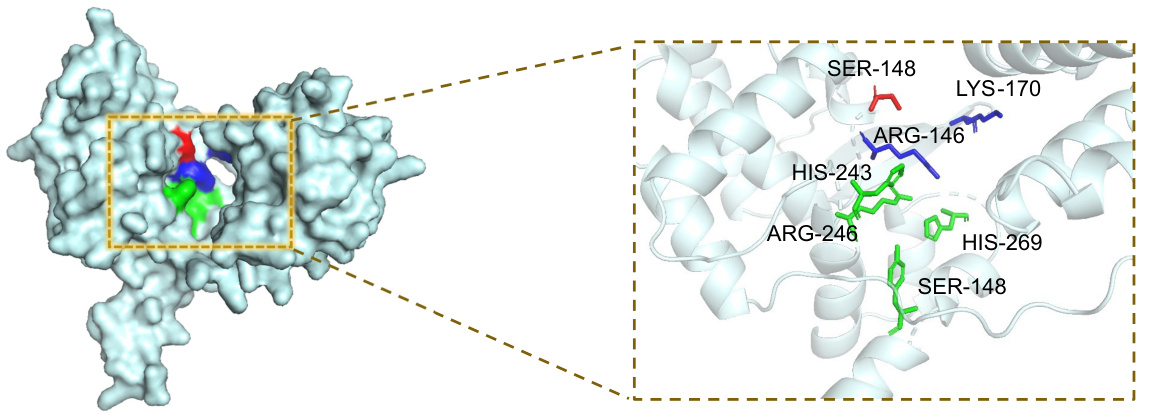
This figure visualizes the results of active site prediction for the protein Tyrosine recombinase XerC using the MMSite framework. The 3D structure of the protein is shown, with the amino acid residues predicted as active sites highlighted in green, incorrectly predicted sites in red, and unpredicted sites in blue. This visualization helps to understand the model’s performance by showing which residues were correctly, incorrectly, or missed by the model’s prediction.
This figure visualizes the results of active site prediction for the protein Tyrosine recombinase XerC using the MMSite framework. The 3D structure of the protein is shown, with amino acid residues colored according to their prediction status: green for correctly predicted active sites, blue for unpredicted active sites, and red for incorrectly predicted sites. This allows for a visual comparison of the model’s predictions against the actual active sites.

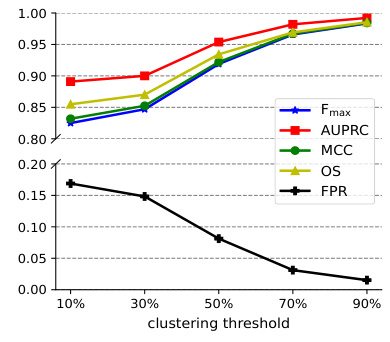
This figure shows the performance of MMSite model on token and region level evaluation metrics (average Fmax, AUPRC, MCC, OS and FPR) when changing the hyperparameter τ from 0.2 to 2.0. The vertical dashed line represents τ=0.8 which is the optimal value for this hyperparameter, according to the paper.

This figure provides a detailed overview of the MMSite framework’s architecture, illustrating the three stages involved in identifying active sites in proteins. It shows how the framework integrates both protein sequence and multi-attribute textual descriptions using a combination of pre-trained models (PLM and BLM), manual prompting, a multi-attribute cross-attention module (MACross), soft-label alignment, and multi-modal fusion. The figure also highlights the ‘First Align, Then Fuse’ strategy and the role of an agent model during inference.
This figure provides a detailed overview of the MMSite framework’s architecture, illustrating the three stages involved: feature extraction, semantic alignment, and multi-modal fusion for active site prediction. It highlights the use of PLMs and BLMs, the MACross module for processing multi-attribute descriptions, and the ‘First Align, Then Fuse’ strategy for integrating the different modalities. The figure also notes the inference stage where a text generation agent model handles missing textual descriptions.
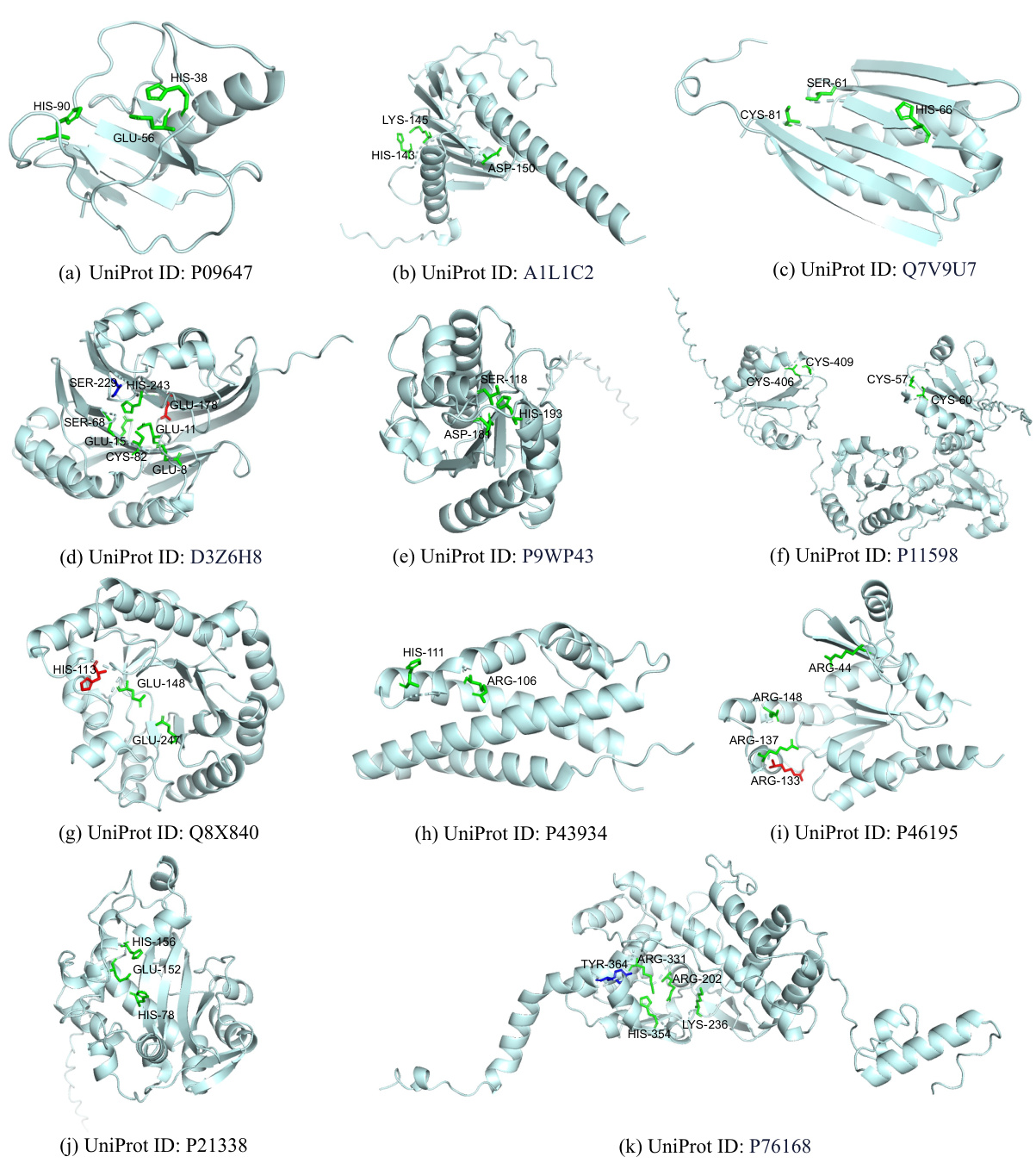
This figure visualizes the results of active site prediction for the protein Tyrosine recombinase XerC. The protein structure is shown, with amino acid residues colored to indicate whether they were correctly predicted as active sites (green), incorrectly predicted (red), or not predicted (blue). This helps illustrate the accuracy and performance of the MMSite model.
More on tables
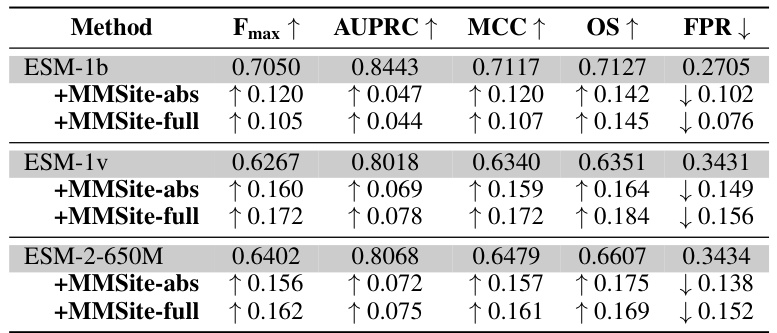
This table presents a comparison of the MMSite model’s performance against 21 other protein representation learning (PRL) models. The comparison is based on a dataset with a clustering threshold of 10%, meaning that protein sequences with more than 10% sequence similarity were grouped together and only one representative sequence from each group was selected. The table shows various metrics to evaluate the performance of each model, including the Fmax, AUPRC, MCC, OS, and FPR. These metrics assess aspects such as the accuracy, precision, and recall of the models in identifying active sites in proteins, considering both sequence-only and multi-modal approaches. The input modalities used by the models are also indicated (Sequence, Structure, or both). The standard deviation (2σ) is included to show the variability in the results.
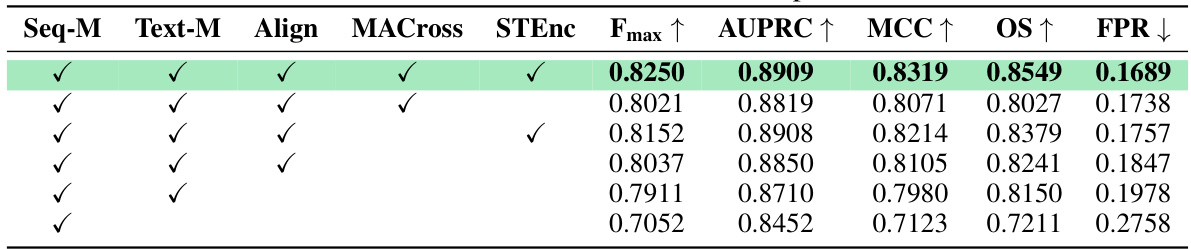
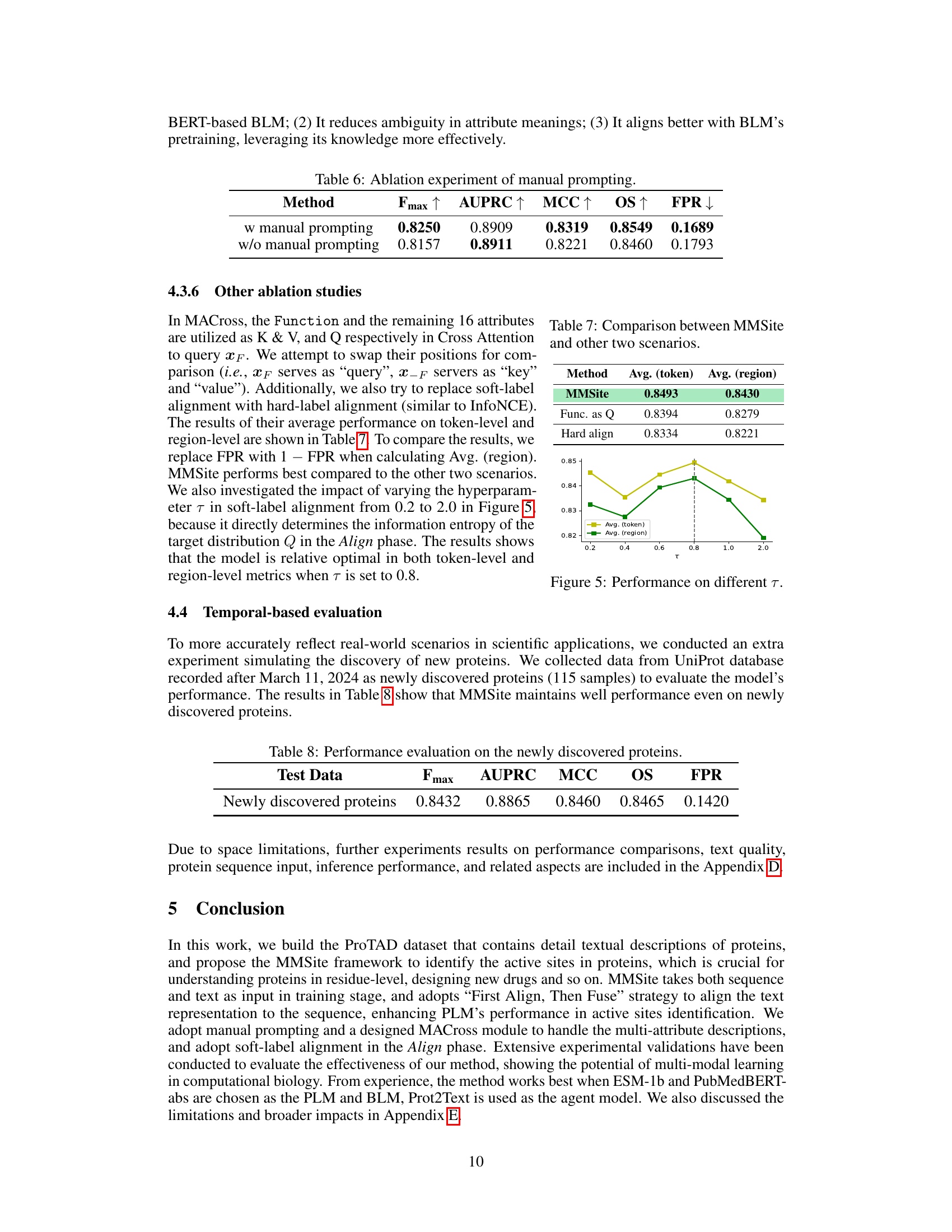
This table presents the ablation study results for the MMSite framework. It shows the impact of removing each component (Seq-M, Text-M, Align, MACross, STEnc) on the overall performance, as measured by Fmax, AUPRC, MCC, OS, and FPR. The row with all components checked represents the full MMSite model’s performance. By comparing this row to the rows with one or more components removed, we understand the individual and combined contributions of each component to the final performance.
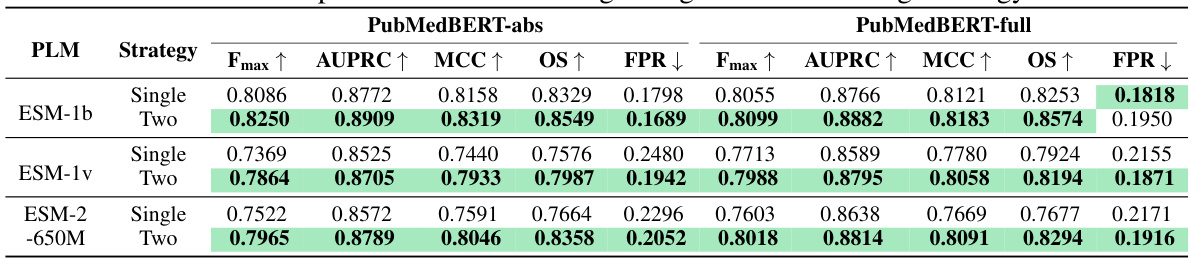
This table presents a comparison of the MMSite model’s performance against 21 other protein representation learning (PRL) models on a dataset with a 10% clustering threshold. The table shows the Fmax, AUPRC, MCC, OS, and FPR metrics for each model. These metrics evaluate the accuracy of the models in identifying active sites within protein sequences. The table also specifies whether the input modality for each model is sequence only, structure only, or a combination of sequence and structure. The results are presented as mean values ± 2 standard deviations, reflecting the variability in performance across multiple trials.
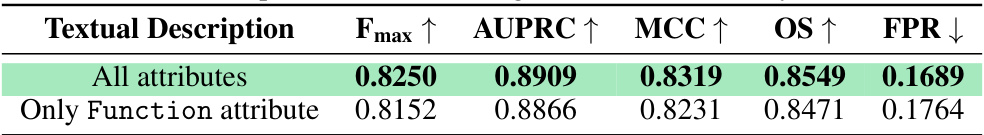
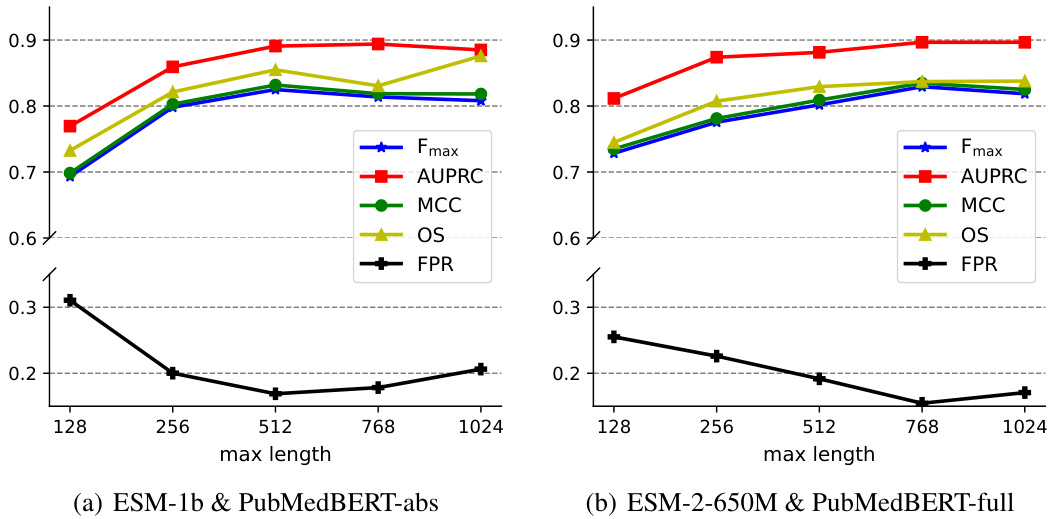
This table presents the performance comparison of the MMSite model using two different approaches: one using all 17 attributes in the textual descriptions and another using only the ‘Function’ attribute. The comparison is made across multiple evaluation metrics, including Fmax, AUPRC, MCC, OS (Overlap Score), and FPR (False Positive Rate). The results demonstrate the impact of incorporating various attributes versus focusing solely on the ‘Function’ attribute for protein active site prediction.

This table presents the ablation study results for MMSite, showing the impact of different components on the model’s performance. It compares the full model’s performance against versions where key components (soft-label alignment, MACross module, shared transformer encoder, text modality) are removed. This helps to understand the contribution of each component to the overall performance improvement.

This table compares the average performance of MMSite against two alternative approaches. The first alternative, labeled ‘Func. as Q’, changes the input order in the multi-modal fusion process. The second alternative uses hard-label alignment instead of the soft-label alignment in MMSite. Both token-level (average precision and recall) and region-level metrics are reported and compared across the three methods. The results demonstrate the effectiveness of the MMSite approach.

This table presents the performance comparison of the proposed MMSite model against 21 other protein representation learning (PRL) models on a dataset with a clustering threshold of 10%. The comparison is done using various metrics (Fmax, AUPRC, MCC, OS, and FPR), all reported as the mean ± 2 standard deviations. The table also indicates whether the input modality used by each model was sequence (Seq.), structure (Struct.), or a combination of both (Seq. & Struct.), as well as the specific model version.

This table compares the performance of MMSite against 21 other protein representation learning (PRL) models on a dataset where the clustering threshold is set to 10%. The comparison considers various metrics like Fmax, AUPRC, MCC, OS, and FPR to evaluate the effectiveness of the models. The table includes the input modality used by each model (sequence, structure, or both) and highlights MMSite’s superior performance.
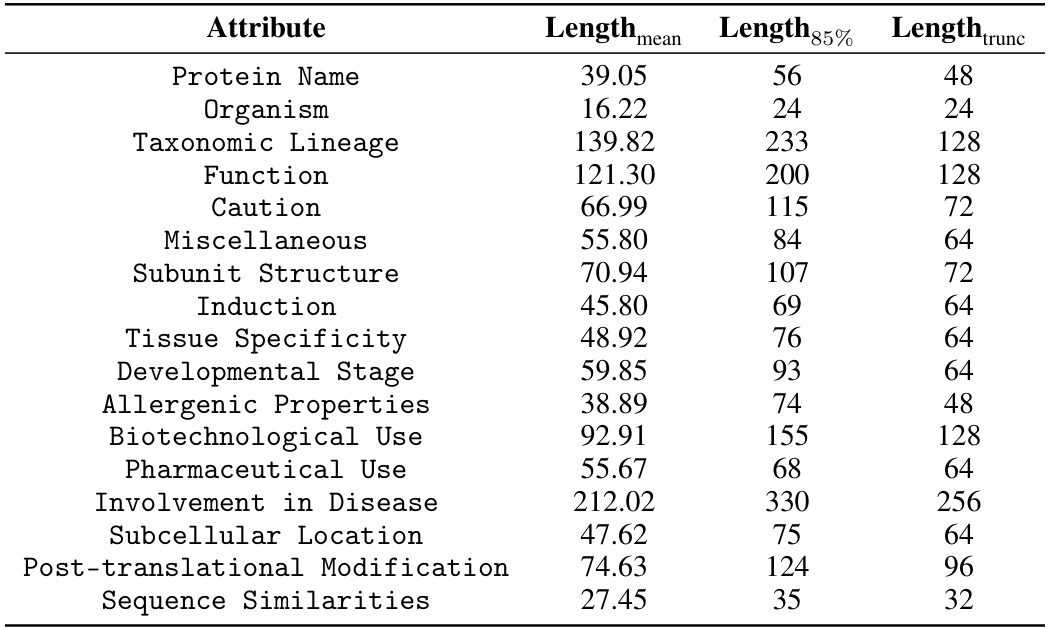
This table presents a comparison of the MMSite framework’s performance against 21 other protein representation learning (PRL) models. The comparison is conducted on a dataset with a clustering threshold of 10%, and the results are presented in terms of several metrics (Fmax, AUPRC, MCC, OS, FPR), all of which are averaged over multiple runs and reported with a standard deviation of two times the standard error. Abbreviations are provided for Seq. (sequence) and Struct. (structure) input types.

This table presents a comparison of the MMSite model’s performance against 21 other protein representation learning (PRL) models. The comparison is based on a dataset where the clustering threshold is set at 10%. The table shows performance metrics (Fmax, AUPRC, MCC, OS, and FPR) for each model, categorized by the input modality (sequence only, structure only, or both) and highlighting the superior performance of MMSite. All values represent the mean ± 2 standard deviations.

This table presents a comparison of the MMSite model’s performance against 21 other protein representation learning (PRL) models on a dataset with a 10% clustering threshold. The performance is evaluated using several metrics, including Fmax, AUPRC, MCC, OS, and FPR. Different input modalities (sequence only, sequence and structure, sequence and text) are considered for the comparison. The table shows that MMSite achieves state-of-the-art performance across the metrics, surpassing models that leverage only sequence or sequence and structure information.

This table compares the performance of MMSite against 21 other protein representation learning (PRL) models on a dataset where the clustering threshold is set to 10%. The metrics used for comparison include Fmax, AUPRC, MCC, OS (Overlap Score), and FPR (False Positive Rate) which are calculated at the residue level. The input modalities of each model (Sequence only, Sequence and Structure, Sequence and Text) are specified, along with the specific version or architecture used for each model. The results are presented as mean values with a 2-standard deviation error bars (±2σ). Abbreviations used in the table are also explained.
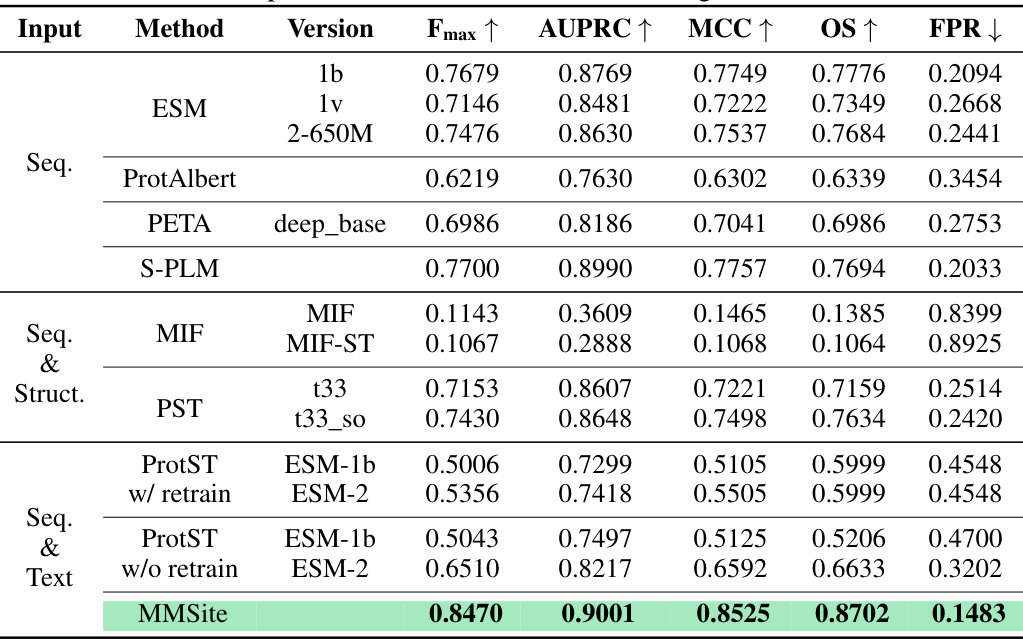
This table presents a comparison of the MMSite model’s performance against 21 other protein representation learning (PRL) models on a dataset with a clustering threshold of 10%. The comparison uses several metrics to evaluate performance: Fmax (F-measure), AUPRC (Area Under the Precision-Recall Curve), MCC (Matthews Correlation Coefficient), OS (Overlap Score), and FPR (False Positive Rate). The input modalities used by each model are indicated, showing whether only sequences (Seq.), structures (Struct.), or a combination of both (Seq. & Struct.) were utilized for prediction. The table helps illustrate the superior performance of MMSite compared to existing state-of-the-art PRL models in the task of protein active site identification.

This table presents a comparison of the MMSite model’s performance against 21 other protein representation learning (PRL) models. The comparison is performed on a dataset where the clustering threshold is set to 10%. The table shows the performance metrics of each model, including Fmax, AUPRC, MCC, OS (Overlap Score), and FPR (False Positive Rate). The input modalities for each model are also specified (Sequence, Structure, Sequence & Structure, or Sequence & Text). The results are presented as the mean value plus or minus two standard deviations. Abbreviations for Sequence and Structure are also provided.

This table presents a comparison of the MMSite model’s performance against 21 other protein representation learning (PRL) models. The comparison is done on a dataset with a clustering threshold of 10%, focusing on the identification of protein active sites. The table shows multiple performance metrics (Fmax, AUPRC, MCC, OS, FPR) for each model, indicating its effectiveness in predicting active sites. The input modality used by each model (sequence only, structure only, or both) is also specified.
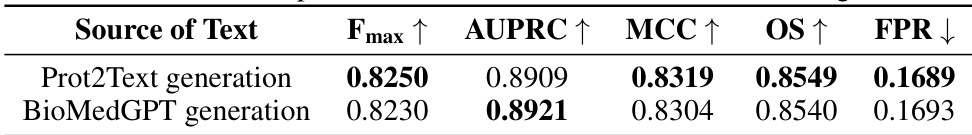
This table compares the performance of MMSite with 21 other protein representation learning (PRL) models on a dataset. The performance is evaluated using several metrics including Fmax, AUPRC, MCC, OS, and FPR. Different input modalities (sequence only, sequence and structure, sequence and text) are considered for each model. The results show that MMSite outperforms all other methods, especially when compared to models that only use protein sequences as input.
Full paper#