↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Current methods in computational biology for understanding gene expression are limited by their focus on single sequence modalities (DNA, RNA, or protein). These approaches fail to capture the intricate interplay between different biological sequences, hindering accurate predictions, especially regarding the complex process of RNA alternative splicing and isoform generation. This complexity leads to difficulties in predicting the expression levels of different RNA isoforms that arise from a single gene, across diverse human tissues. This is crucial for understanding disease mechanisms and gene regulation.
To address these limitations, this paper introduces IsoFormer, a novel multi-modal model designed to integrate DNA, RNA, and protein sequence data to predict RNA transcript isoform expression levels. IsoFormer’s innovative architecture leverages pre-trained modality-specific encoders to effectively transfer knowledge across different biological sequences. This methodology proved highly successful, outperforming existing single-modality approaches. The study demonstrates efficient transfer learning within and between modalities, highlighting the significant benefit of this integrative approach for solving complex biological problems. The researchers also open-sourced their model, IsoFormer, to facilitate further research and development in the field.
Key Takeaways#
Why does it matter?#
This paper is highly significant for researchers in bioinformatics and computational biology due to its novel multi-modal approach to gene expression prediction. It addresses the critical need for more comprehensive models capable of handling the complexity of biological data. The open-sourced model and associated code will accelerate further research in this area, particularly in the investigation of alternative splicing and its role in disease. The findings are highly relevant to current trends in foundation models and transfer learning, paving the way for new multi-modal gene expression approaches.
Visual Insights#

This figure illustrates the architecture of the IsoFormer model’s aggregation module. The module takes as input modality-specific embeddings from DNA, RNA, and protein sequences, generated by pre-trained encoders. It uses cross-attention layers to integrate information from different modalities and residual connections to improve information flow. This multi-modal embedding is then passed to a task-specific head to generate the final prediction, enabling the model to effectively leverage information from all three sequence modalities.

This table lists the characteristics of the different single-modality encoders used in the IsoFormer model. For each modality (DNA, RNA, and Protein), it shows the specific model used, the type of pre-training employed (Supervised or Self-Supervised), the number of parameters in the model, the tokenization scheme (how many nucleotides or amino acids constitute a single token), and the maximum sequence length that the model can handle. This table provides essential information for understanding the foundation models upon which the IsoFormer architecture is built.
In-depth insights#
Multimodal Gene Expression#
Multimodal gene expression analysis holds significant promise for a deeper understanding of gene regulation. By integrating data from various sources like DNA sequence, RNA expression levels, and protein abundance, researchers can move beyond the limitations of single-modality approaches. A multimodal approach allows for the capture of complex interactions and regulatory mechanisms that cannot be observed by examining a single data type. This holistic perspective is crucial for understanding how genes respond to environmental changes, developmental cues, and disease states. The integration of different data modalities allows for more robust and accurate predictions of gene expression patterns in various tissues and cell types, paving the way for more precise diagnostics and targeted therapies. However, challenges remain in the development of computationally efficient and scalable methods for handling the high-dimensional, heterogeneous data inherent in multimodal analysis. The lack of sufficient standardized datasets and the computational costs associated with advanced machine learning models also need to be addressed to fully realize the potential of multimodal gene expression analysis.
IsoFormer Architecture#
The IsoFormer architecture is a novel multi-modal approach designed for efficient integration of DNA, RNA, and protein sequence information. It leverages pre-trained modality-specific encoders to generate embeddings for each sequence type. These embeddings are then processed through an aggregation module, which could use various techniques like cross-attention, to create a unified multi-modal representation. This unified representation is finally fed into a task-specific head, in this case for RNA transcript isoform expression prediction. The architecture’s strength lies in its ability to effectively capture the interconnectedness of biological sequences and leverage the knowledge embedded within the pre-trained models. The use of pre-trained encoders allows for efficient transfer learning, both within and between modalities. Flexibility is incorporated by allowing the choice of different encoders and aggregation methods, making the architecture adaptable to a range of biological tasks.
Transfer Learning Effects#
In exploring transfer learning effects within the context of biological foundation models, a key consideration is how effectively pre-trained models on single-modality data (DNA, RNA, or protein) can generalize to multi-modal tasks. The success of transfer learning hinges on the shared representational features across modalities. If the pre-trained models capture fundamental biological principles, then transferring this knowledge to a multi-modal model should improve performance significantly. However, differences in data distributions and inherent complexities of biological processes may limit the extent of successful transfer. Careful consideration of the aggregation strategy which combines the multi-modality embeddings becomes crucial; inefficient aggregation would hinder the benefits of transfer learning. Furthermore, the selection of appropriate pre-trained models and their suitability for the downstream task are essential for maximizing transfer learning gains. Investigating various encoder architectures and exploring different aggregation mechanisms, alongside comprehensive evaluation metrics, would offer a more nuanced understanding of transfer learning’s impact.
Aggregation Strategies#
The effectiveness of multi-modal learning hinges significantly on the strategy employed for aggregating information from diverse modalities. The paper explores various aggregation strategies, comparing their performance in a multi-modal biological sequence model. Cross-attention, a method allowing modalities to interact and learn from each other directly, emerges as a top performer. However, the study also investigates alternatives like the Perceiver Resampler and C-Abstractor, which aim to reduce computational costs or enhance representation learning. While these alternatives offer different trade-offs between efficiency and accuracy, the results highlight the strength and interpretability of cross-attention in this specific application, where understanding the interplay between modalities is crucial. The choice of aggregation strategy is shown to be non-trivial and significantly impact the model’s overall performance in predicting RNA transcript isoform expression.
Future Research#
Future research directions stemming from this multi-modal biological sequence modeling work could focus on several key areas. Expanding the number of modalities integrated into the model, such as incorporating epigenetic data or 3D structural information, could significantly improve prediction accuracy. Exploring alternative aggregation methods beyond the current cross-attention approach would enhance the model’s ability to capture complex relationships between biological sequences. Applying the model to a wider range of biological tasks is crucial, including predicting gene regulation, protein-protein interactions, and disease susceptibility. Furthermore, developing more efficient and scalable training methods would allow the analysis of larger and more complex datasets. Finally, investigating the model’s robustness to noise and incomplete data is critical, as real-world biological data are often imperfect. Addressing these research avenues will significantly advance our understanding of biological systems and pave the way for the development of more sophisticated biological foundation models.
More visual insights#
More on figures

This figure illustrates the architecture of the aggregation module used in the IsoFormer model. The module takes as input the embeddings from three modality-specific encoders (DNA, RNA, and protein) and uses cross-attention layers to combine information from these modalities. The cross-attention layers allow the model to learn relationships between the different modalities, and the residual connections help to improve the model’s accuracy. The output of the aggregation module is a multi-modal embedding that can be used to predict the desired output.

Figure 2(a) illustrates the central dogma of molecular biology, showing how DNA is transcribed into RNA, which is then translated into proteins. It highlights that a single DNA sequence can produce multiple RNA isoforms due to alternative splicing, and these isoforms can lead to different protein isoforms. Figure 2(b) is a schematic of the IsoFormer model. It shows how pre-trained encoders for DNA, RNA, and protein sequences generate embeddings which are combined by an aggregation module to create multi-modal embeddings. These embeddings then predict the expression levels of each RNA isoform in multiple human tissues.
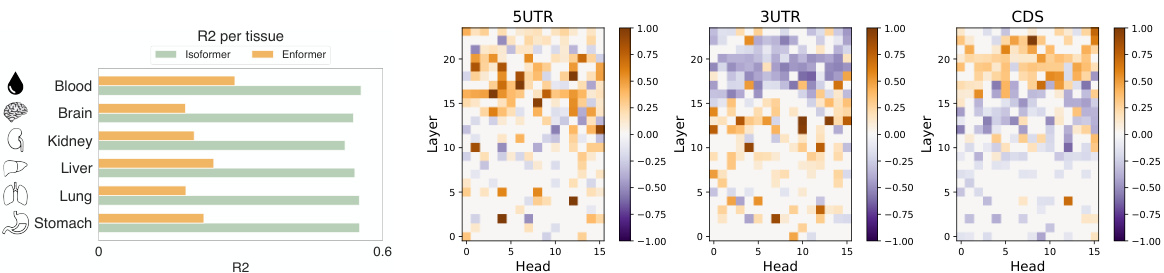
Figure 2a illustrates the central dogma of molecular biology, showing how DNA is transcribed into RNA, which is then translated into proteins. A single gene can produce multiple RNA isoforms (due to alternative splicing) that translate into different protein isoforms. The abundance of each isoform varies across different tissues. Figure 2b depicts the IsoFormer model’s architecture. This multi-modal model takes DNA, RNA, and protein sequences as input, uses pre-trained encoders for each modality to generate embeddings, and aggregates these embeddings using an aggregation module. A prediction head then uses this multi-modal representation to predict the expression level of each RNA isoform across tissues.

This figure compares four different aggregation strategies used in the IsoFormer model for combining information from DNA, RNA, and protein modalities. Each strategy is visually represented showing its architecture, including cross-attention, resamplers, and linear projections. The goal is to show how these different approaches integrate the information from different modalities to create a single, multi-modal representation. The figure focuses on the creation of the multi-modal DNA embedding (h’dna), but the same principles apply to the RNA and protein embeddings.

Figure 2(a) illustrates the central dogma of molecular biology, highlighting the interconnectedness of DNA, RNA, and protein sequences in gene expression. It demonstrates how a single gene can produce multiple RNA isoforms and protein isoforms due to alternative splicing. Figure 2(b) shows the architecture of the IsoFormer model, which utilizes pre-trained modality-specific encoders (for DNA, RNA, and proteins) to generate modality-specific embeddings. These embeddings are then aggregated into multi-modal embeddings, which are used to predict the expression levels of RNA transcripts across multiple tissues.
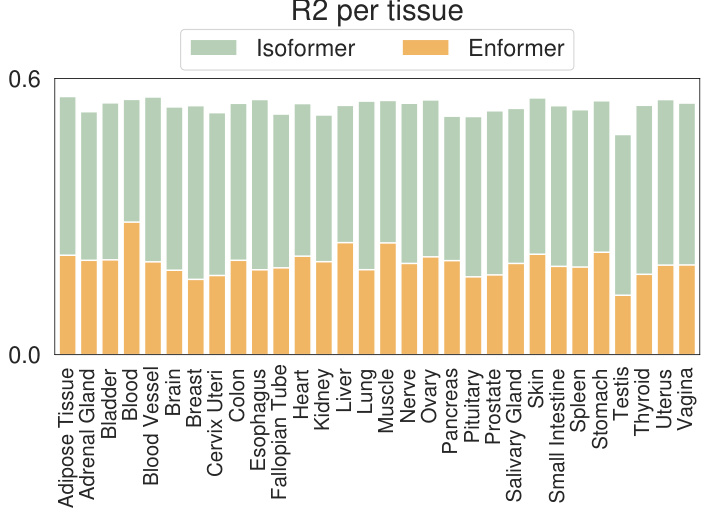
This figure is a bar chart comparing the performance of IsoFormer and Enformer models for RNA transcript isoform expression prediction across 30 different human tissues. Each bar represents a tissue, and the height of the bar shows the R-squared (R2) value, a measure of how well the model predicts expression levels. The bars are segmented, with the bottom portion showing Enformer’s R2 and the top portion showing the improvement achieved by IsoFormer. The chart visually demonstrates IsoFormer’s superior performance in most tissues.
More on tables

This table presents the performance of IsoFormer using different combinations of input modalities (DNA, RNA, and protein) for the task of transcript isoform expression prediction. The results are shown in terms of R-squared (R2) and Spearman correlation, both calculated across multiple tissues and averaged across five independent random trials. The table demonstrates how the inclusion of additional modalities improves the prediction performance.

This table compares the performance of IsoFormer using different DNA encoders: Enformer and Nucleotide Transformer (NT). It shows the R-squared (R²) and Spearman correlation values across 30 human tissues for the transcript isoform expression prediction task. The results are averaged across five different random seeds to provide a measure of variability. The table helps demonstrate the impact of the choice of DNA encoder on IsoFormer’s performance.
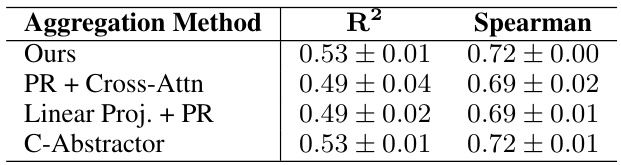
This table presents the results of an ablation study comparing four different aggregation methods used in the IsoFormer model for integrating information from DNA, RNA, and protein sequences. The methods are compared based on the R-squared (R²) and Spearman correlation values obtained for predicting RNA transcript isoform expression. The table shows that the proposed aggregation method (‘Ours’) performs as well or better than the alternative approaches. The DNA encoder used across all methods is Enformer.
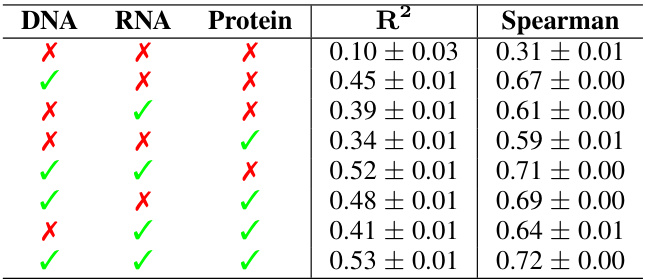
This table shows the performance of IsoFormer when using different combinations of pre-trained and randomly initialized encoders for DNA, RNA, and protein modalities. The results (R-squared and Spearman correlation) demonstrate the impact of pre-trained encoders on the model’s performance, highlighting the effectiveness of transfer learning from pre-trained models.
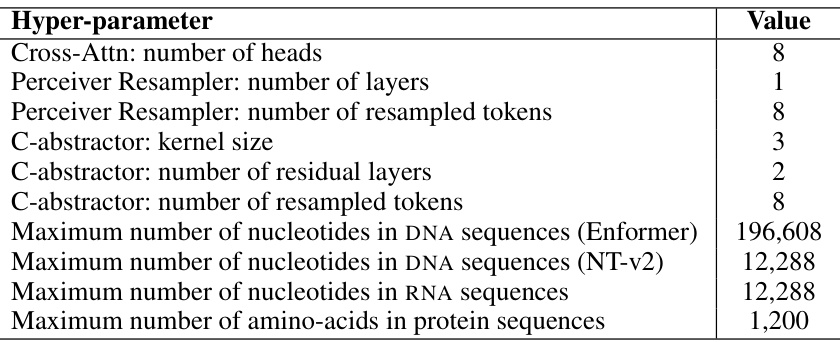
This table lists the hyperparameters used in the IsoFormer model. It includes settings for different components such as the cross-attention mechanism, the Perceiver Resampler, and the C-abstractor. The maximum sequence lengths for the DNA, RNA, and protein sequences are also given, reflecting the input limitations of the respective encoders (Enformer and Nucleotide Transformer).
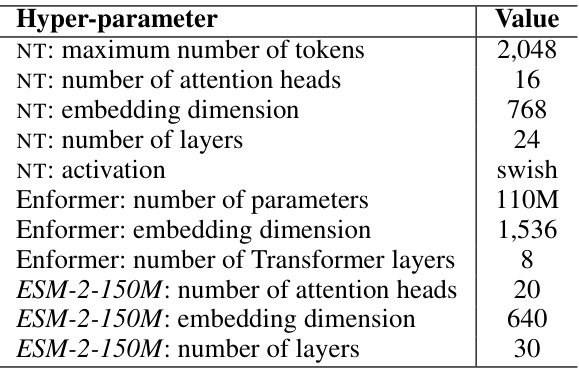
This table lists the hyperparameters used for the three pre-trained encoders used in the IsoFormer model. These hyperparameters are not trained but taken from the original pre-trained models. They include details such as the maximum number of tokens, number of attention heads, embedding dimensions, and number of layers for each of the DNA (NT and Enformer), RNA (NT), and protein (ESM-2-150M) encoders. The activation function used is also specified.

This table presents the performance of IsoFormer model variants with different combinations of input modalities (DNA, RNA, Protein) for predicting transcript isoform expression. The performance is evaluated using R-squared (R2) and Spearman correlation across multiple human tissues, averaged over five runs with different random seeds. The results demonstrate the impact of incorporating multiple modalities on model accuracy.
Full paper#



















