↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Aptamer discovery, crucial in drug development, relies heavily on high-throughput screening—a time-consuming and costly process. Current machine learning methods struggle due to a scarcity of labeled training data. This limitation makes it hard to accurately predict the binding of aptamers to proteins. This research aims to overcome these challenges by developing an unsupervised method for predicting these interactions.
The researchers introduce FAFormer, a novel deep learning model based on a transformer architecture. This model leverages predicted pairwise contact maps between proteins and nucleic acids to estimate binding affinity. FAFormer achieves higher accuracy in contact map prediction than previous methods and demonstrates exceptional speed, making it suitable for large-scale analysis. In real-world tests with several protein-aptamer datasets, FAFormer proves highly effective at identifying strong binding candidates, thereby offering a significant advancement in aptamer screening.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in drug discovery and computational biology. It offers a novel unsupervised method for protein-nucleic acid interaction prediction, significantly accelerating aptamer screening, which is currently a time-consuming and costly process. The proposed FAFormer model presents a new architecture with potential implications for other geometric deep learning applications. Its superior speed and performance also address the computational limitations of existing models, allowing researchers to work with larger and more complex datasets.
Visual Insights#
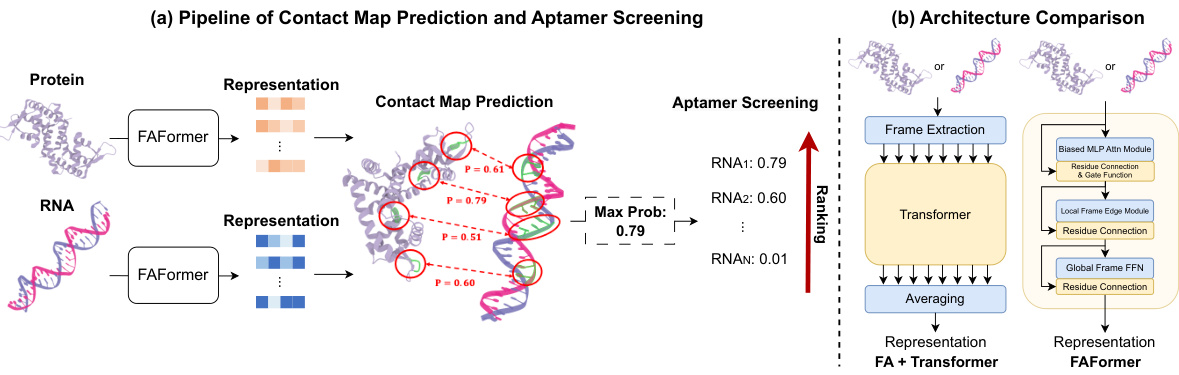
This figure illustrates two aspects of the FAFormer model. (a) shows the overall workflow: the model predicts contact maps between proteins and nucleic acids, and uses the maximum contact probability to estimate binding affinity for aptamer screening. (b) contrasts FAFormer’s architecture with a standard transformer using frame averaging, highlighting FAFormer’s unique integration of frame averaging into each transformer block for enhanced geometric information processing.
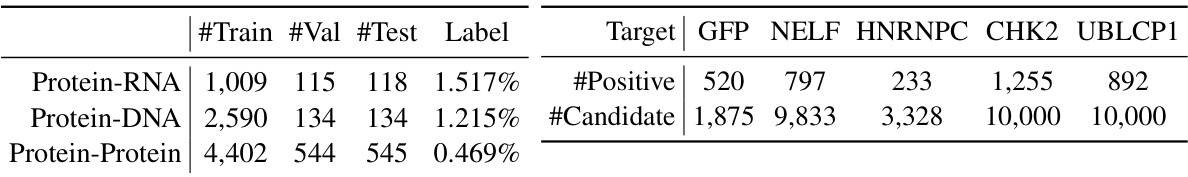
The table presents statistics for three protein complex datasets used in the study. Each row represents a dataset (Protein-RNA, Protein-DNA, Protein-Protein). The columns indicate the number of training, validation, and test samples in each dataset, along with the percentage of positive labels (indicating the presence of a contact). This information is crucial for understanding the size and characteristics of the datasets used to train and evaluate the models.
In-depth insights#
FAFormer: A Deep Dive#
FAFormer, as the name suggests, is a novel deep learning architecture built upon the Transformer model. Its core innovation lies in seamlessly integrating frame averaging (FA) within each Transformer block. This is a significant departure from standard FA methods, which typically treat FA as a separate preprocessing step. By integrating FA directly into the Transformer blocks, FAFormer effectively infuses geometric information into node features while preserving the spatial semantics of coordinates. This results in a model with increased expressive power and better performance in tasks involving geometric data. The architecture’s design, featuring a Local Frame Edge Module, a Biased MLP Attention Module, and a Global Frame FFN, enables sophisticated processing of spatial relationships between nodes. This is particularly relevant for protein-nucleic acid complex modeling, which demands the accurate representation and understanding of geometric structures. FAFormer’s ability to learn equivariant transformations for symmetry groups, a key challenge in geometric deep learning, offers significant advantages in handling the inherent symmetries found in molecules. The results from experiments indicate that FAFormer significantly outperforms existing methods, achieving over a 10% relative improvement in contact map prediction and demonstrating the effectiveness of this approach in large-scale aptamer screening.
Equivariant Modeling#
Equivariant modeling in the context of this research paper is crucial for effectively capturing the inherent symmetries present in protein-nucleic acid complexes. Standard machine learning models often struggle with such data because they don’t inherently understand rotational or translational invariance. Equivariant models address this by incorporating geometric information directly into their architecture, enabling them to learn representations that remain consistent regardless of the molecule’s orientation in space. This is particularly important when analyzing binding interactions, as the relative positions of residues and nucleotides are key to understanding affinity. The paper likely explores several techniques to achieve equivariance, perhaps employing methods such as frame averaging or group convolution. The use of an equivariant framework leads to more robust and accurate predictions, especially valuable in the context of unsupervised learning where labeled data is scarce. This allows the model to generalize better to unseen protein-nucleic acid complexes.
Contact Map Prediction#
The task of contact map prediction in protein-nucleic acid complex modeling presents a significant challenge, demanding sophisticated methods to accurately predict pairwise interactions. The accuracy of such predictions is crucial for understanding the binding affinity and subsequently, for applications like aptamer screening. This involves computationally intensive methods for complex structure prediction. The study leverages unsupervised learning approaches, avoiding the limitations of relying on scarce labeled datasets. A key aspect is the model’s ability to integrate geometric information into node features while preserving spatial semantics, which is vital for accurate contact map prediction. This task is benchmarked against several state-of-the-art equivariant models, demonstrating superior performance with a notable relative improvement. The predicted contact maps serve as a strong indicator for aptamer screening, effectively identifying high-affinity binding candidates, highlighting the model’s applicability for accelerating the drug discovery pipeline.
Aptamer Screening#
Aptamer screening, a crucial step in discovering novel therapeutic agents, is traditionally a time-consuming and expensive process. This research proposes a significant advancement by leveraging machine learning to predict protein-nucleic acid interactions, thereby enabling unsupervised aptamer screening. Instead of relying on costly and laborious experimental methods, the model predicts contact maps between proteins and nucleic acids. The maximum probability from these maps serves as a strong indicator of binding affinity, allowing for the efficient ranking and selection of promising aptamer candidates. This approach demonstrates a potential paradigm shift in aptamer discovery, offering a substantial speedup and reduced cost compared to traditional methods while achieving comparable performance. The implications are far-reaching, potentially accelerating the development of novel therapeutics and diagnostics based on nucleic acid-based drugs.
Future Directions#
Future research could explore enhancing FAFormer’s capabilities by incorporating more sophisticated geometric features, such as dihedral angles and torsion angles, to better capture the three-dimensional structure of protein-nucleic acid complexes. Improving the handling of long-range interactions within the Transformer architecture remains crucial, potentially through the use of more advanced attention mechanisms. Expanding the model’s applicability to other types of biomolecular interactions, like protein-protein or protein-small molecule interactions, would broaden its impact. Addressing the issue of data scarcity in the field is essential, and exploring techniques such as data augmentation or transfer learning could significantly improve the model’s generalizability. Finally, investigating the biological mechanisms underlying protein-nucleic acid interactions, as revealed by the contact maps predicted by FAFormer, could lead to a deeper understanding of biological processes and inspire novel drug discovery strategies.
More visual insights#
More on figures

This figure provides a detailed illustration of the FAFormer architecture, highlighting its key components: Biased MLP Attention Module, Local Frame Edge Module, Global Frame FFN, and Gate Function. It showcases how the input node features, coordinates, and edge representations are processed through a stack of these modules, illustrating the integration of frame averaging (FA) within the Transformer architecture. The figure also emphasizes the distinction between local and global frames in capturing both local and long-range interactions within the molecular structure.
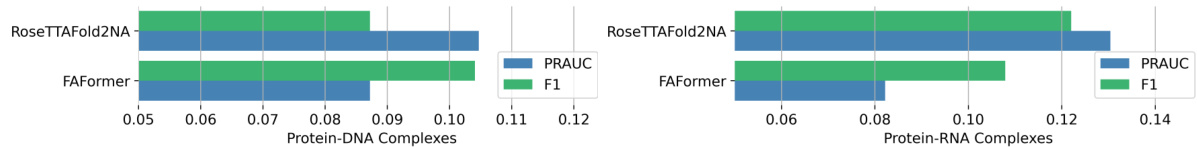
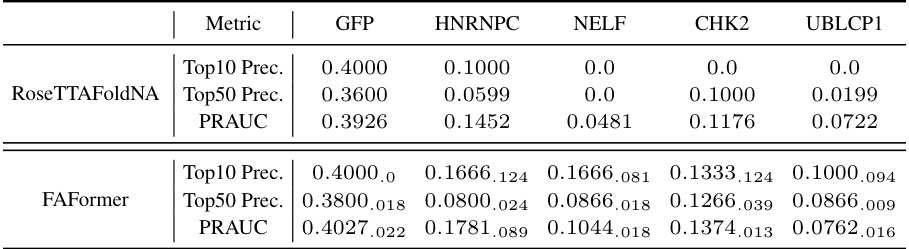
This figure compares the performance of FAFormer and RoseTTAFoldNA on contact map prediction using the test sets from RoseTTAFoldNA. It shows the F1 score and PRAUC (Precision-Recall Area Under the Curve) for both models, separately for protein-DNA and protein-RNA complexes. The bar chart visually represents the performance difference, highlighting FAFormer’s competitive performance relative to RoseTTAFoldNA, especially considering that FAFormer does not utilize Multiple Sequence Alignments (MSAs), unlike RoseTTAFoldNA.

This figure shows a comparison of ground truth and FAFormer predicted contact maps for two protein-nucleic acid complexes (7DVV and 7KX9). Heatmaps visualize the contact probability between each residue and nucleotide. The left side of each row shows the actual contact map, and the right side displays FAFormer’s prediction. This visual comparison demonstrates FAFormer’s ability to accurately predict contact maps, even with sparse contact pairs.
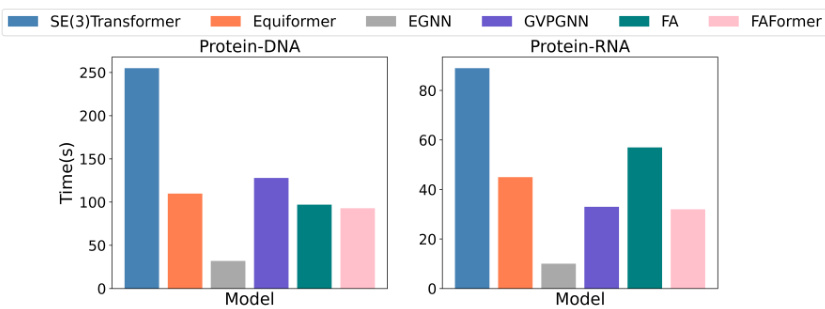
This figure compares the training time of FAFormer against other baseline models (SE(3)Transformer, Equiformer, EGNN, GVP-GNN, and FA) for protein-DNA and protein-RNA datasets. It visually represents the efficiency of FAFormer in terms of training time compared to these other models. The bar chart shows the training time in seconds for each model on each dataset.
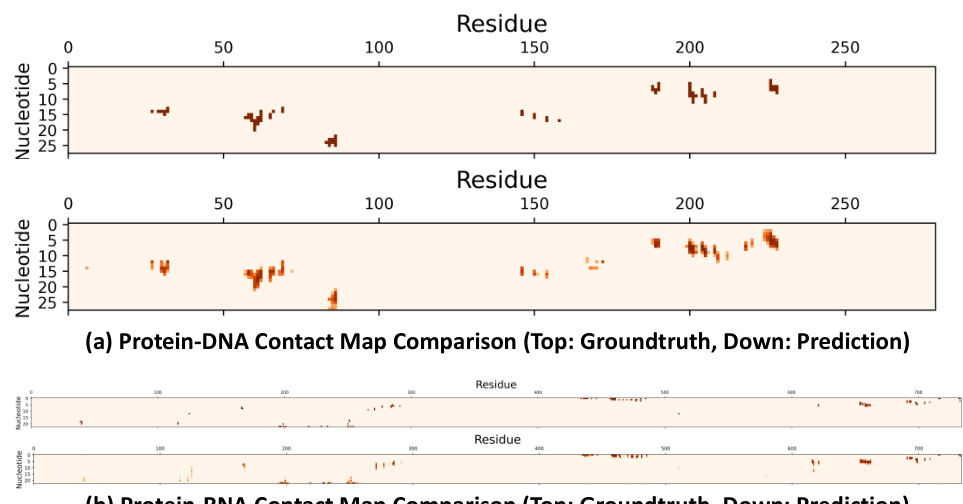
This figure shows a comparison of ground truth and predicted contact maps for two protein-nucleic acid complexes (7DVV and 7KX9). The heatmaps visualize the contact probability between each nucleotide and residue in the complexes. The top row in each panel shows the ground truth contact map, while the bottom row shows the contact map predicted by the FAFormer model. The figure demonstrates the model’s ability to accurately capture the sparse patterns of contacts in these protein-nucleic acid complexes.
More on tables

This table presents the comparison of FAFormer’s performance against other baseline methods on three protein complex datasets (Protein-RNA, Protein-DNA, and Protein-Protein) for the task of contact map prediction. The metrics used for evaluation are F1 score and PRAUC (Precision-Recall Area Under the Curve). The table shows FAFormer consistently outperforms other models across all datasets, demonstrating its effectiveness in this task.
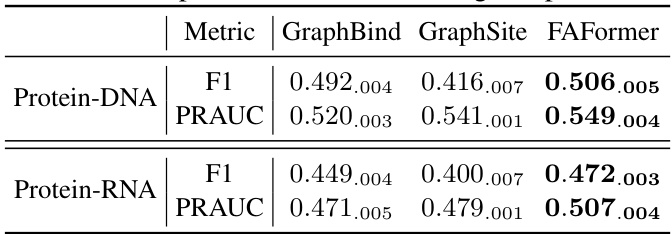
This table presents the comparison of the performance of FAFormer against two state-of-the-art geometric deep learning models, GraphBind and GraphSite, on the task of binding site prediction. The metrics used for comparison are F1 score and PRAUC (Precision-Recall Area Under the Curve). Results are shown separately for Protein-DNA and Protein-RNA datasets, indicating the model’s effectiveness in identifying nucleic-acid-binding residues on proteins.
This table presents the comparison of the performance of FAFormer against other baseline methods for the contact map prediction task. It shows the F1 score and PRAUC (Precision-Recall Area Under the Curve) values across three different protein complex datasets: Protein-RNA, Protein-DNA, and Protein-Protein. The results highlight FAFormer’s improved performance compared to other methods, indicating its effectiveness in predicting protein-nucleic acid interactions.
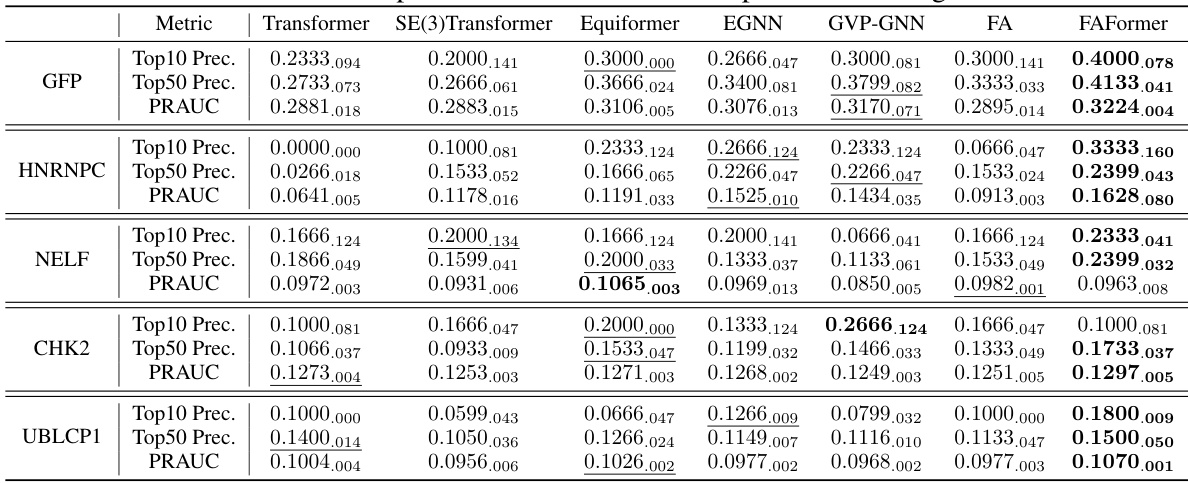
This table compares the performance of FAFormer and RoseTTAFoldNA on the aptamer screening task using sampled datasets. It shows the top 10 precision, top 50 precision, and PRAUC (Precision-Recall Area Under the Curve) for each of five protein targets (GFP, NELF, HNRNPC, CHK2, and UBLCP1). The sampled datasets are smaller subsets of the original datasets, intended to account for the computational cost differences between the two methods. The results highlight the comparative performance of FAFormer, indicating its effectiveness even with reduced dataset sizes.

This table compares the average and total inference times of RoseTTAFoldNA and FAFormer for the contact map prediction task. The times include the time taken to predict unbound structures for FAFormer. The comparison highlights the significantly faster inference speed of FAFormer, particularly noticeable when considering the time required for generating unbound protein and RNA structures.
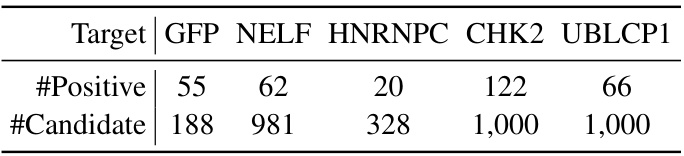
This table presents the statistics of the five sampled aptamer datasets used in the comparison between RoseTTAFoldNA and FAFormer. For each target protein (GFP, NELF, HNRNPC, CHK2, UBLCP1), the number of positive aptamers and the total number of candidate aptamers are shown. The sampled datasets were created by randomly selecting 10% of the candidates from the original datasets.

This table presents a comparison of the performance of FAFormer against several baseline methods on three different protein complex datasets (Protein-RNA, Protein-DNA, and Protein-Protein). The performance metrics used are F1 score and PRAUC (Precision-Recall Area Under the Curve). The results demonstrate that FAFormer outperforms all other methods on all three datasets.

This table compares the performance of AlphaFold3, RoseTTAFoldNA, and FAFormer on two aptamer datasets (GFP and HNRNPC) in terms of Top10 Precision, Top50 Precision, and PRAUC. The results show that FAFormer outperforms AlphaFold3 in all metrics and shows comparable results to RoseTTAFoldNA.
Full paper#



















