↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Dual-target drug design, aiming to create single drugs effective against two targets simultaneously, is challenging due to data scarcity and complex interactions. Current methods often struggle with generating molecules that effectively bind to both targets. This research focuses on overcoming these obstacles using AI.
The study introduces a novel dataset based on synergistic drug combinations. It proposes a novel approach to repurpose pretrained diffusion models designed for single-target drugs to generate dual-target drugs. The approach leverages SE(3)-equivariant message passing and compositional drift to guide the generation process, effectively transferring knowledge gained during single-target training to the dual-target scenario. This zero-shot learning approach is shown to be highly effective, surpassing various strong baselines and demonstrating significant improvements in the design of dual-target drugs.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in drug design due to its novel dataset of synergistic drug combinations and innovative approach using reprogrammed diffusion models for dual-target drug discovery. It offers a zero-shot transfer learning method, overcoming data scarcity issues, and provides strong baselines for comparison. This opens avenues for further research using AI to enhance dual-target drug design, potentially improving drug efficacy and reducing resistance.
Visual Insights#
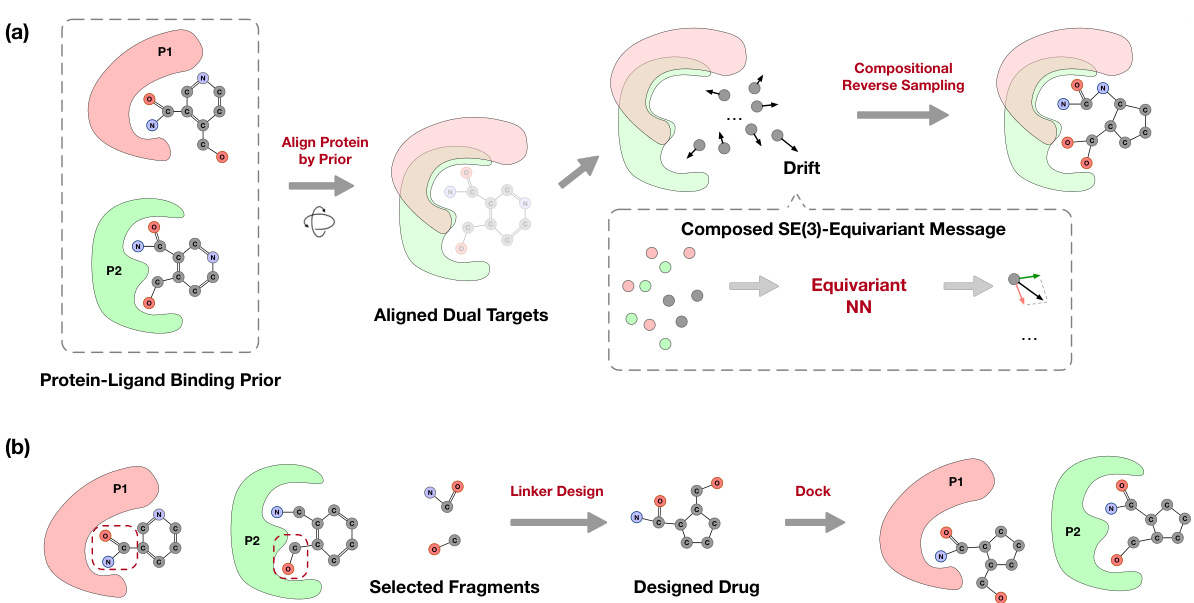
This figure illustrates the two main methods proposed in the paper for dual-target drug design. (a) shows the COMPDIFF and DUALDIFF methods, which involve aligning dual protein pockets using protein-ligand binding priors, constructing complex graphs, and employing SE(3)-equivariant message passing to generate dual-target drugs via compositional reverse sampling. (b) demonstrates the repurposing of linker design methods, where fragments from reference molecules are identified, linked using linker design methods, and docked to form a complete dual-target drug molecule.
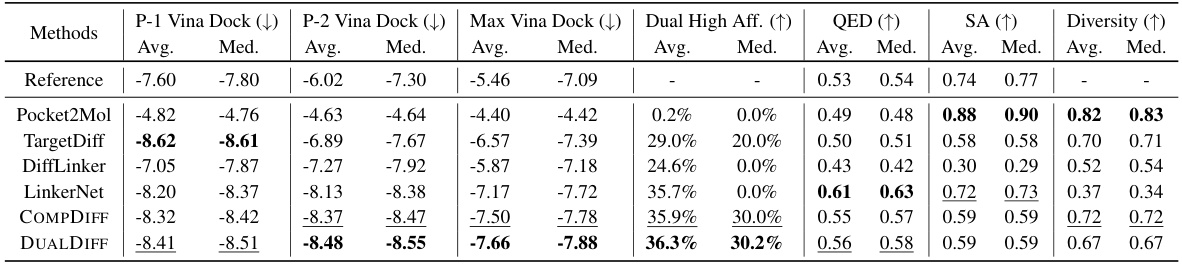
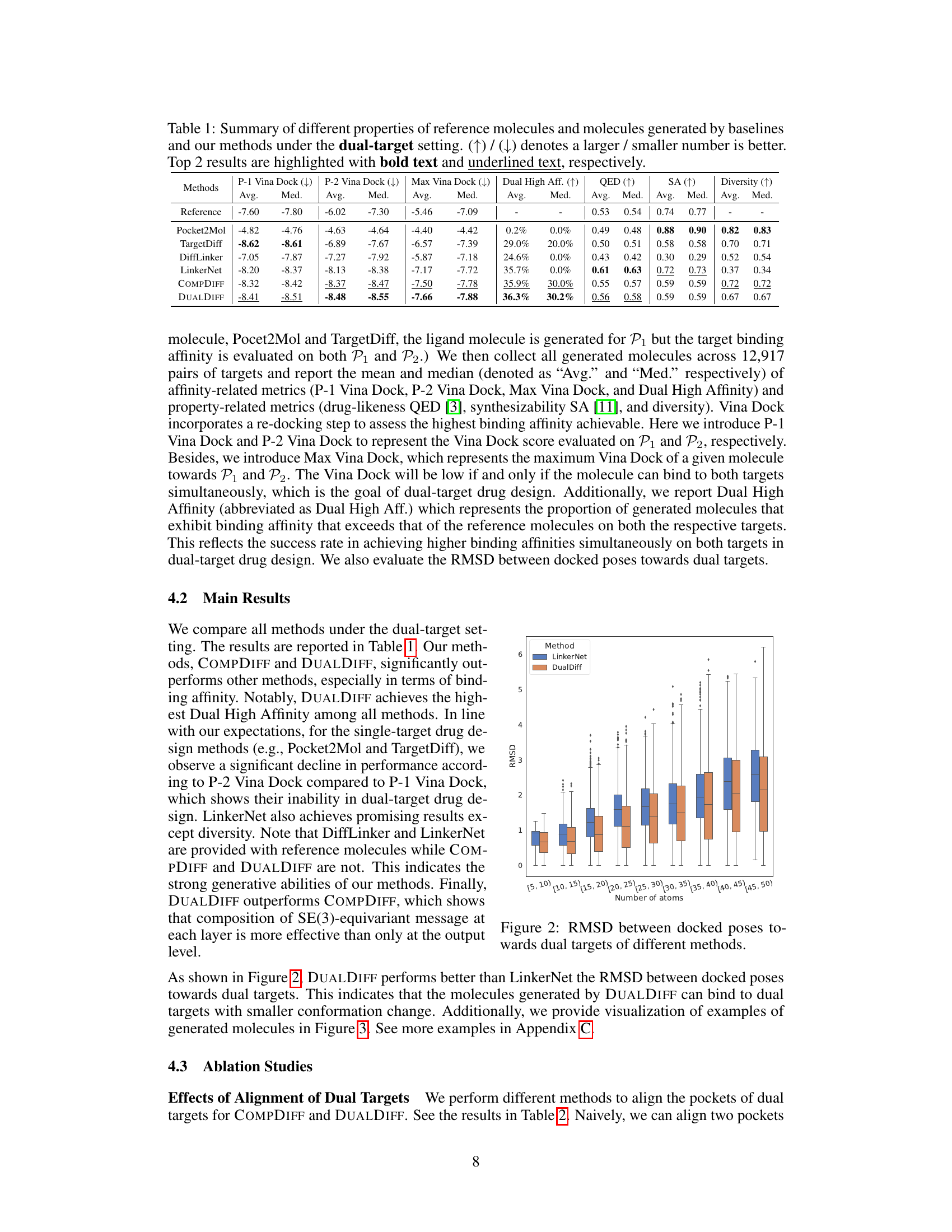
This table compares the performance of different methods for dual-target drug design. It shows various properties of reference molecules and those generated by different methods, including binding affinity to each target (P-1 Vina Dock, P-2 Vina Dock, Max Vina Dock), the percentage of molecules with higher binding affinity than the reference (Dual High Aff.), and other properties like drug-likeness (QED), synthesizability (SA), and diversity. Higher values are generally better for most metrics except for Vina Dock scores.
In-depth insights#
Dual-Target Drug Design#
Dual-target drug design represents a paradigm shift in pharmaceutical research, aiming to develop single molecules capable of interacting with two distinct biological targets simultaneously. This approach offers several advantages over single-target strategies, including the potential to overcome drug resistance, enhance therapeutic efficacy, and reduce side effects by addressing multiple disease pathways. The key challenge in this area lies in the complex interplay of factors influencing target selection, molecule design, and drug synergy, demanding sophisticated computational tools and experimental validation. Deep generative models and geometric deep learning have emerged as promising avenues for accelerating this process, enabling the generation of novel molecules with desirable properties. Successful dual-target drug design hinges on identifying synergistic target pairs, designing molecules with complementary interactions, and carefully characterizing their effects in preclinical studies, ultimately leading to more effective and safer treatments.
Diffusion Model Reprogramming#
Diffusion model reprogramming is a powerful technique that leverages the capabilities of pretrained diffusion models for new tasks, avoiding the need for extensive retraining. This is achieved by cleverly manipulating the model’s components, such as the drift or message passing mechanisms, to adapt the model to a different yet related task. The method is particularly effective when dealing with limited data for the new target task, as it effectively transfers knowledge from the source task to the target task. In the context of dual-target drug design, this approach is particularly relevant, as generating compounds that interact with two distinct targets simultaneously is challenging due to the lack of readily available training data. By reprogramming a pretrained model, the approach cleverly overcomes this data scarcity limitation. The core idea lies in aligning the two target pockets appropriately, then strategically composing the output from separate diffusion processes for the two target pockets, leveraging the insights already learned for single-target interaction. This elegant technique allows for effective knowledge transfer and superior performance, surpassing traditional methods. Furthermore, the method can be extended to other generative tasks. The success of the diffusion model reprogramming approach opens new avenues in generative modeling.
SE(3)-Equivariant Messaging#
SE(3)-equivariant message passing leverages the power of group theory to design neural networks that are inherently invariant to rotations and translations in 3D space. This is particularly valuable in tasks such as molecular design, where the spatial arrangement of atoms is critical but their absolute position is not. By incorporating SE(3) equivariance, the model can learn features that are robust to variations in orientation and position, leading to more accurate and generalizable predictions. This approach is especially beneficial when dealing with limited training data, as it allows the model to generalize better from the available samples. A key advantage is its applicability to dual-target drug design, where aligning two protein pockets requires understanding spatial relationships between ligands and their binding sites. This sophisticated approach allows for better representation and handling of the complex 3D structures involved in drug-protein interactions. The improved robustness and generalization capabilities from SE(3) equivariance are significant for improving the quality and efficacy of generative models in the challenging field of structure-based drug discovery.
Synergistic Drug Combinations#
The concept of synergistic drug combinations is central to this research, driving the dataset curation and shaping the experimental design. The authors leverage the synergistic effect, where the combined effect of two drugs exceeds the sum of their individual effects, to rationally select target pairs for dual-target drug design. This approach is more purposeful than selecting random target pairs, as it is grounded in the clinically observed phenomenon of drug synergism. This methodology allows for the development of a meaningful dataset of potential dual-target drugs with demonstrably improved efficacy and reduced side effects. The selection process also incorporates considerations of in vivo efficacy and cellular-level effects to further enhance the practical relevance of the identified synergistic combinations and ensure the dataset’s usability for real-world applications. The focus on synergism ultimately steers the research towards more effective and targeted drug development.
Future Directions and Limitations#
This research makes significant strides in dual-target drug design using diffusion models, but several avenues for future work exist. Improving the accuracy and diversity of generated molecules is crucial, perhaps through incorporating more sophisticated scoring functions that better capture nuanced protein-ligand interactions. Addressing the challenge of conformational flexibility in proteins during the binding process is also important, as the current model simplifies this aspect. Expanding the dataset to encompass more diverse target pairs and synergistic drug combinations would strengthen the generalizability of the method. Finally, thorough in vivo testing and validation are necessary to confirm the efficacy and safety of the designed drugs before clinical trials. The current focus on in silico methods warrants this crucial next step for translation to practical applications. While the zero-shot transfer learning approach is a notable strength, direct comparisons with fine-tuning strategies on larger dual-target datasets would provide valuable insights into the relative strengths and weaknesses of each approach.
More visual insights#
More on figures
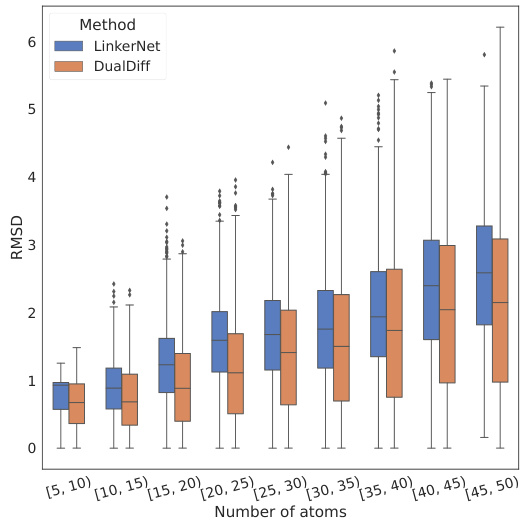
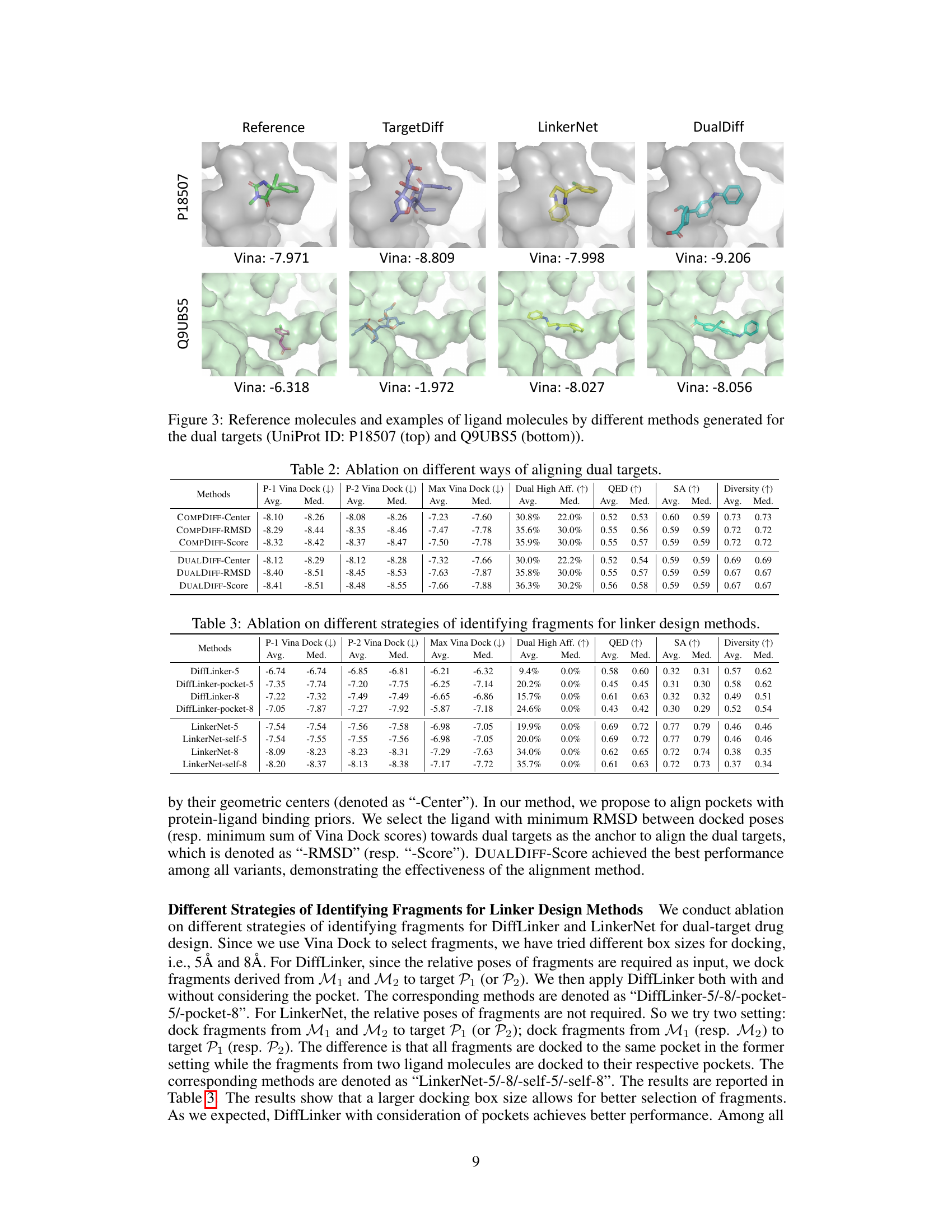
This figure shows the root-mean-square deviation (RMSD) between the docked poses of molecules generated by different methods towards dual targets. The x-axis represents the number of atoms in the molecule, grouped into bins. The y-axis represents the RMSD value. Box plots show the distribution of RMSD values for molecules generated by LinkerNet and DualDiff for each atom count bin. The figure demonstrates that DualDiff tends to produce molecules with lower RMSDs, indicating better binding affinity and potentially fewer structural changes during binding to both target proteins.
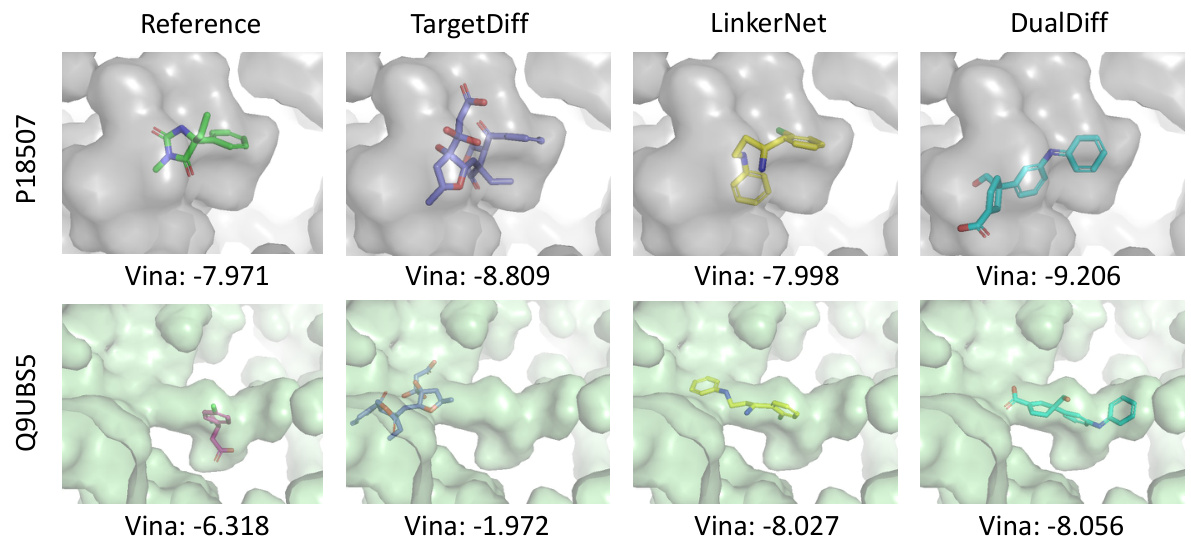
This figure illustrates the methodology used for dual-target drug design. Panel (a) details the COMPDIFF and DUALDIFF methods, emphasizing the alignment of dual pockets using protein-ligand binding priors, the construction of complex graphs, and the generation of dual-target ligands via compositional reverse sampling. Panel (b) shows how linker design methods are repurposed to connect fragments from the reference molecules for dual targets, leading to complete molecules capable of binding to both targets.
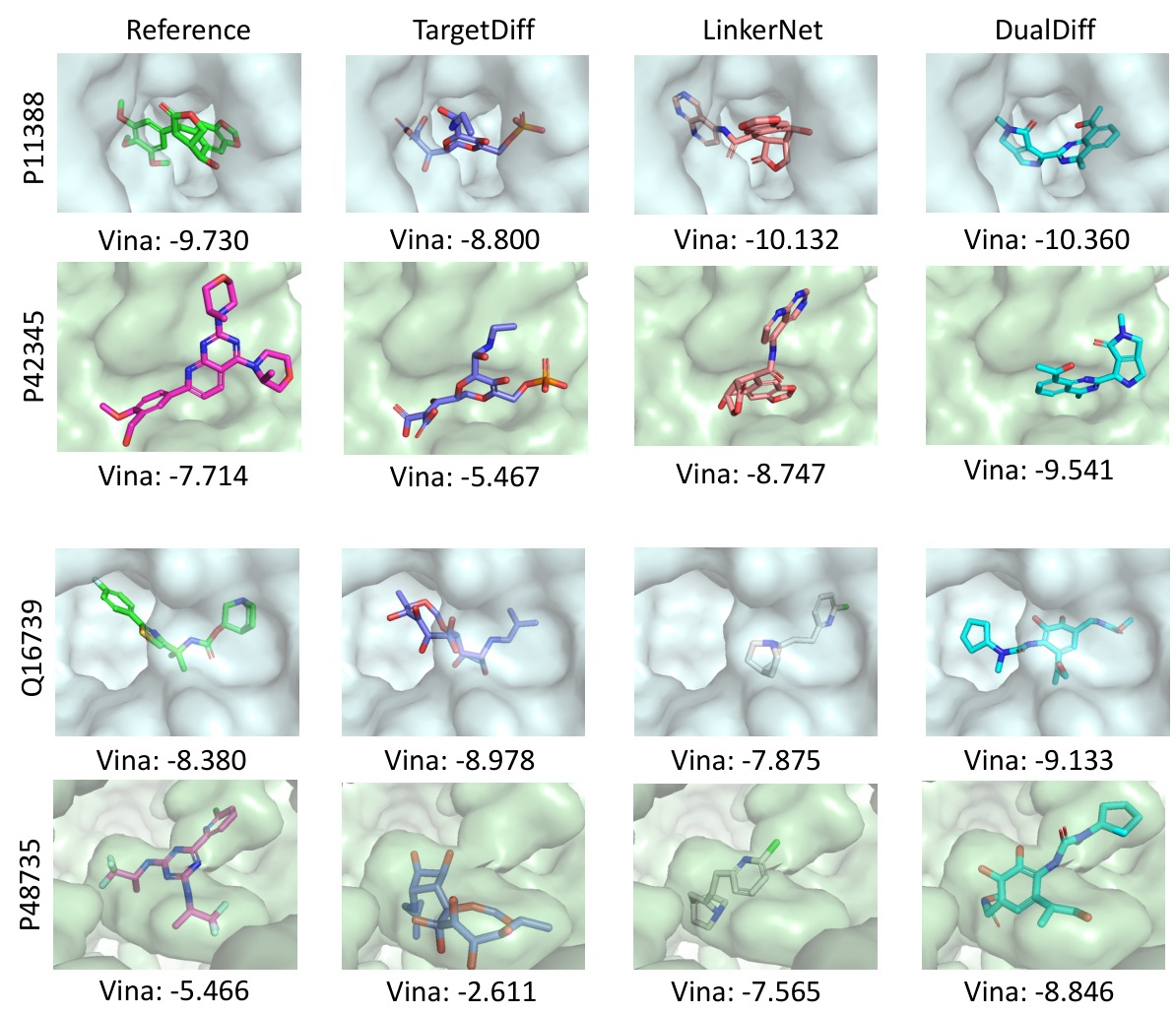
This figure illustrates the proposed method for dual-target drug design. Part (a) shows the COMPDIFF and DUALDIFF methods, which align dual protein pockets using protein-ligand binding priors, create complex graphs, and use SE(3)-equivariant message passing to generate dual-target ligands via compositional reverse sampling. Part (b) details how linker design methods are repurposed by identifying fragments from reference molecules, linking them, and creating molecules that bind to both targets.
More on tables
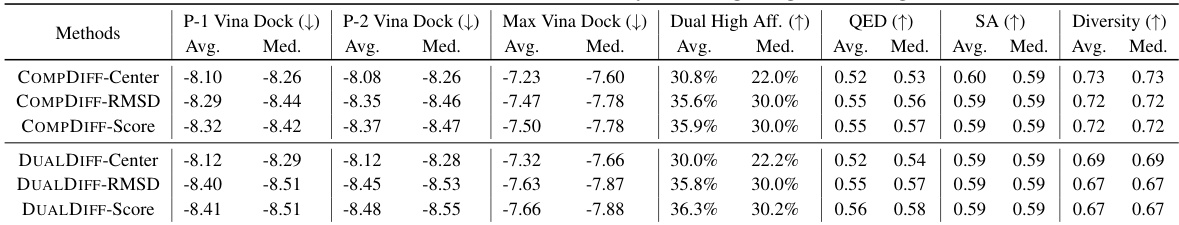
This table summarizes the performance of different methods (including baselines and the proposed methods) for dual-target drug design. It compares various properties of the generated molecules against reference molecules, including binding affinities to the primary and secondary targets (P-1 Vina Dock, P-2 Vina Dock, Max Vina Dock), the percentage of molecules achieving higher binding affinity than the references in both targets (Dual High Aff.), and measures of drug-likeness (QED, SA, and Diversity). Higher values are better for QED, SA, Dual High Aff, and Diversity, while lower values are better for docking scores. The table helps to assess the effectiveness of different methods in generating molecules suitable for dual-target drug design.
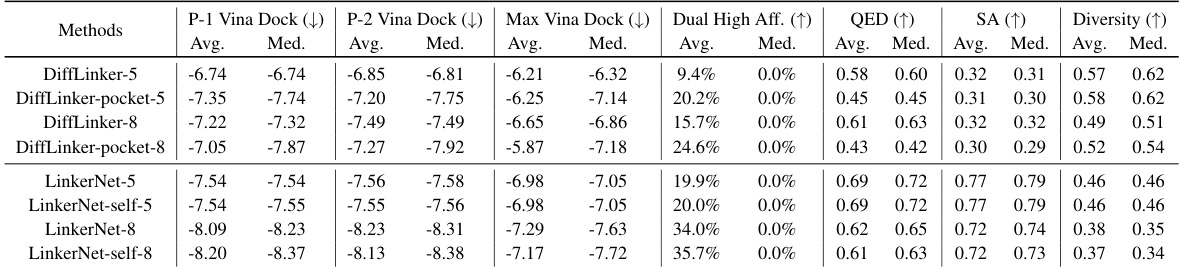
This table summarizes the performance of various methods (baselines and the proposed methods) in dual-target drug design. It compares several key properties of the generated molecules against those of reference molecules, including binding affinity to each target (P-1 Vina Dock, P-2 Vina Dock), the highest binding affinity across both targets (Max Vina Dock), the percentage of generated molecules exceeding reference molecule affinity on both targets (Dual High Aff.), drug-likeness (QED), synthesizability (SA), and molecular diversity. Higher scores generally indicate better performance, with the direction specified by the arrows (↑ or ↓).
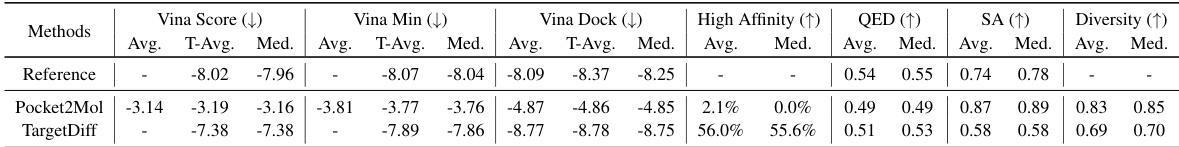
This table presents the performance comparison of reference molecules and molecules generated by two baseline methods (Pocket2Mol and TargetDiff) under the single-target drug design setting. The metrics evaluated include Vina Score (binding affinity), Vina Min (minimum energy after minimization), Vina Dock (highest binding affinity after re-docking), High Affinity (proportion of molecules with higher affinity than the reference molecule), QED (drug-likeness score), SA (synthesizability score), and Diversity. The table highlights the performance differences between the baseline models and the reference molecules in terms of these relevant properties.
Full paper#