↗ OpenReview ↗ NeurIPS Homepage ↗ Chat
TL;DR#
Many protein mutations remain unexplored, despite their significance in biology and medicine. Existing protein language models (PLMs) struggle to provide understandable explanations for mutational effects or design new mutations with specific properties due to their implicit treatment of mutations and limited supervision. They often focus on evolutionary plausibility rather than human-defined criteria. This necessitates explainable and engineerable tools for mutation studies.
MutaPLM addresses these issues. This unified framework uses a novel protein delta network to explicitly model mutations and incorporates chain-of-thought (CoT) learning to extract knowledge from biomedical literature. A new, large-scale dataset, MutaDescribe, provides cross-modal supervision for this model. The results demonstrate MutaPLM’s superior performance in providing human-understandable explanations for mutation effects and prioritizing mutations with desirable properties. The open-sourced code, model, and data significantly advance research in protein mutation analysis and engineering.
Key Takeaways#
Why does it matter?#
This paper is crucial for researchers in protein engineering and computational biology. MutaPLM offers a novel framework for interpreting and engineering protein mutations, moving beyond limitations of existing protein language models. Its open-source nature and comprehensive dataset accelerate progress in understanding protein evolution and design, opening new avenues for drug discovery, biomaterial development, and disease research.
Visual Insights#

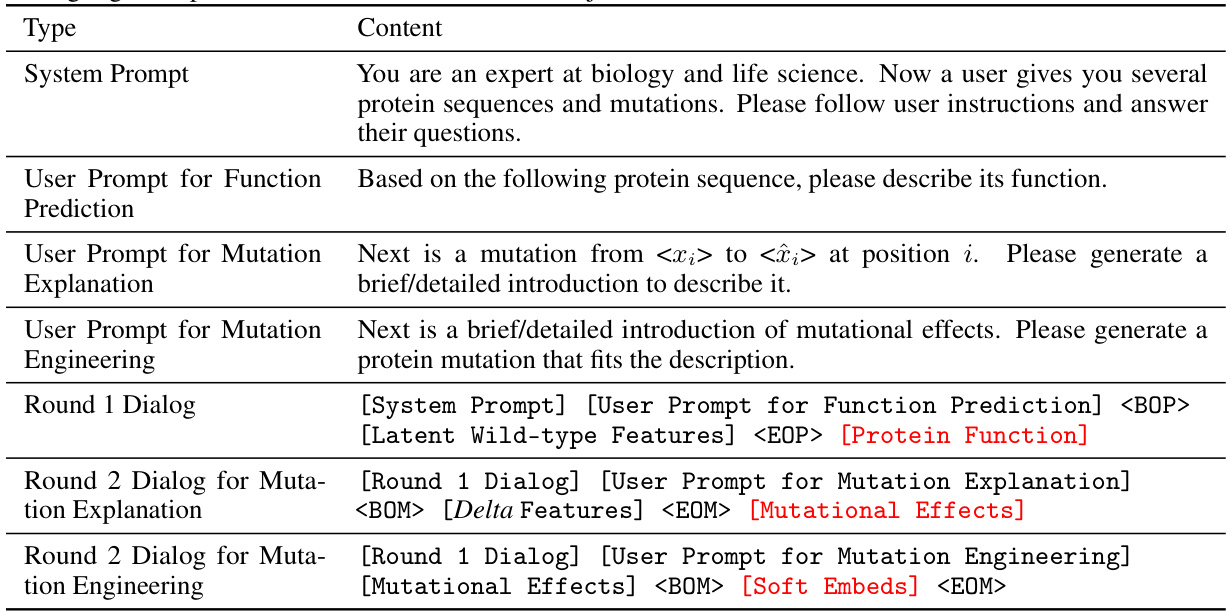
This figure illustrates the architecture of MutaPLM, a framework for interpreting and navigating protein mutations using protein language models. The figure is divided into two parts: (a) the encoding branch and (b) the decoding branch of a protein delta network. The encoding branch takes the difference between the representations of mutant and wild-type proteins (generated by a Protein Language Model, PLM) to create a ‘delta’ representation (z∆). This delta representation captures the essence of the mutation. The decoding branch uses this delta representation, along with the wild-type protein representation and a Large Language Model (LLM), to generate a description of the mutation’s effects and predict the new amino acid sequence. The use of both PLM and LLM allows for cross-modal learning and generation of human-understandable textual explanations.
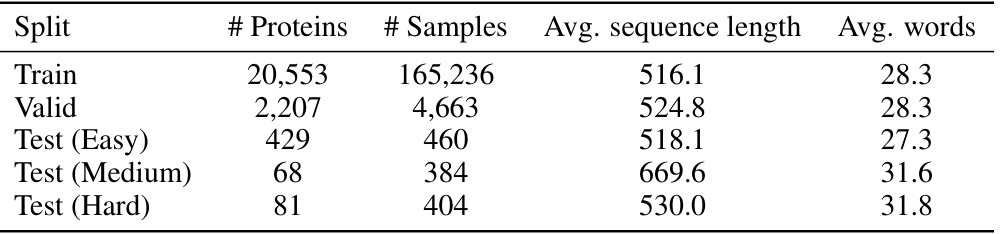
This table presents a statistical overview of the MutaDescribe dataset, a key contribution of the research paper. It shows the number of proteins and samples in the dataset, categorized by the difficulty of the tasks (Train, Valid, and three levels of Test sets). The average length of the protein sequences and average number of words used to describe the mutational effects are also given for each category.
In-depth insights#
MutaPLM Framework#
The MutaPLM framework represents a novel approach to protein mutation analysis and engineering, integrating protein language models (PLMs) and large language models (LLMs). Its core innovation lies in the protein delta network, which explicitly models the difference between wild-type and mutant protein representations, providing a richer feature space for understanding mutational effects. This is complemented by a transfer learning pipeline utilizing chain-of-thought prompting, effectively leveraging biomedical text data to enhance explainability and engineerability. MutaPLM’s explainability stems from its ability to generate human-understandable textual descriptions of mutational effects, going beyond simple fitness scores. Its engineerability is demonstrated by its capacity to prioritize and propose novel mutations with desirable properties, guided by free-text instructions. The framework is further strengthened by the MutaDescribe dataset, a large-scale resource with rich textual annotations of protein mutations, crucial for training and evaluation. In summary, MutaPLM offers a powerful and versatile tool for navigating the complexities of protein mutations, promising significant advancements in protein engineering and biological research.
Delta Network Design#
A thoughtfully designed delta network for protein mutation modeling should explicitly capture the differences between wild-type and mutant protein representations. This could involve using a specialized architecture, perhaps incorporating attention mechanisms to focus on the mutated residues and their surrounding context within the protein sequence. Furthermore, the network needs to handle variable-length protein sequences, as the length of proteins can vary significantly. Effective strategies for achieving this might include using techniques like embedding layers that capture the sequential information of the proteins and pooling mechanisms to generate a fixed-size representation for downstream processing. The design should also allow for transfer learning to leverage pre-trained protein language models effectively. Finally, the network needs to produce a representation that is suitable for subsequent mutation explanation and engineering tasks, potentially using a multimodal approach that combines protein information with other data modalities like textual descriptions of mutational effects.
Transfer Learning#
Transfer learning, in the context of this research paper, is a powerful technique that leverages pre-trained models to improve the efficiency and effectiveness of downstream tasks. Instead of training a model from scratch, the approach uses knowledge gained from a large, general dataset to initialize a model for a more specific, smaller dataset. This is particularly valuable when data for the target task is scarce or expensive to acquire. The study uses transfer learning to extract relevant knowledge about protein mutations from biomedical texts, significantly enriching the model’s understanding beyond the inherent information found in protein sequences alone. This approach addresses the challenges of limited explicit data on mutational effects by leveraging implicit knowledge found in the vast corpus of existing scientific literature. The chain-of-thought prompting method further enhances the effectiveness of transfer learning by guiding the model’s reasoning process, allowing it to better connect abstract information from texts to concrete protein mutation details. Through this combined approach, the model is able to achieve significantly better performance in mutation explanation and engineering tasks than models trained without this knowledge transfer.
MutaDescribe Dataset#
The MutaDescribe dataset represents a significant contribution to the field of protein mutation analysis. Its novelty lies in its large scale, incorporating diverse protein sequences and mutations paired with rich textual annotations describing the effects of those mutations. This cross-modal nature is crucial for training and evaluating explainable and engineerable protein language models (PLMs). The dataset’s structure enables both quantitative and qualitative analyses, moving beyond simple fitness scores to capture the nuanced, human-understandable effects of mutations. By facilitating more comprehensive understanding of mutational impacts, MutaDescribe holds promise for advancing research in various areas, such as protein engineering, drug design, and disease research. The inclusion of detailed textual annotations allows the development of more sophisticated and accurate PLMs capable of explaining complex biological phenomena, thus bridging the gap between in-silico predictions and experimental validation.
Future Directions#
Future research should prioritize expanding the dataset’s scope to encompass more diverse protein mutations and incorporate additional modalities such as protein structure data, enhancing explainability. Integrating active learning could enable the model to leverage experimental feedback, refining its predictions and improving its ability to guide the design of novel, beneficial mutations. Addressing potential biases arising from the training data should be thoroughly investigated. Moreover, exploring the application of MutaPLM to other relevant biological problems (e.g., protein-protein interaction prediction) would demonstrate its versatility. Finally, rigorous safety measures are crucial to prevent misuse; future work should focus on developing mechanisms to ensure the responsible use of the model, particularly in applications related to bioengineering or drug discovery, emphasizing ethical considerations.
More visual insights#
More on figures
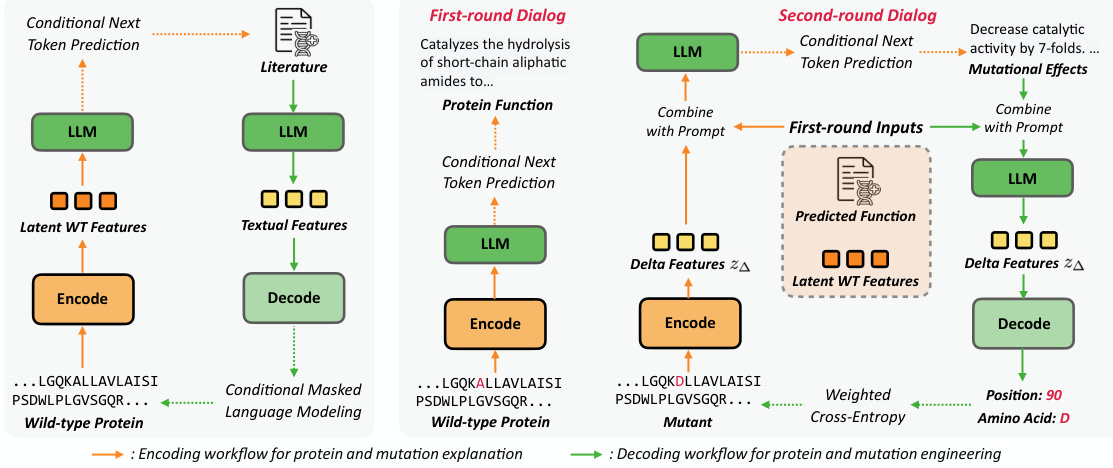
This figure illustrates the training pipeline of the MutaPLM model. Panel (a) shows the pre-training stage on protein literature, where the model learns from both protein sequences and their associated textual descriptions. The encoding workflow uses next token prediction, while the decoding workflow uses conditional masked language modeling. Panel (b) depicts the fine-tuning stage using chain-of-thought prompting. This involves a two-round dialog: first, describing the wild-type protein’s function; second, explaining the mutation’s effects and predicting the mutation based on those effects.
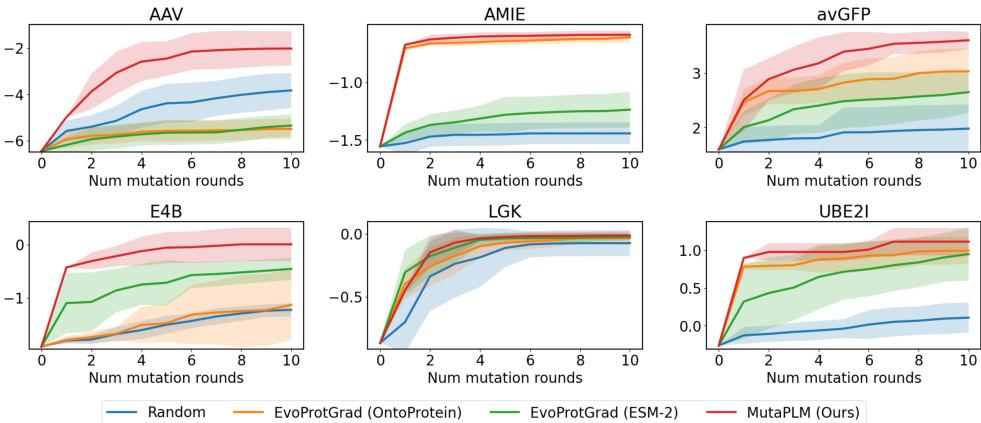
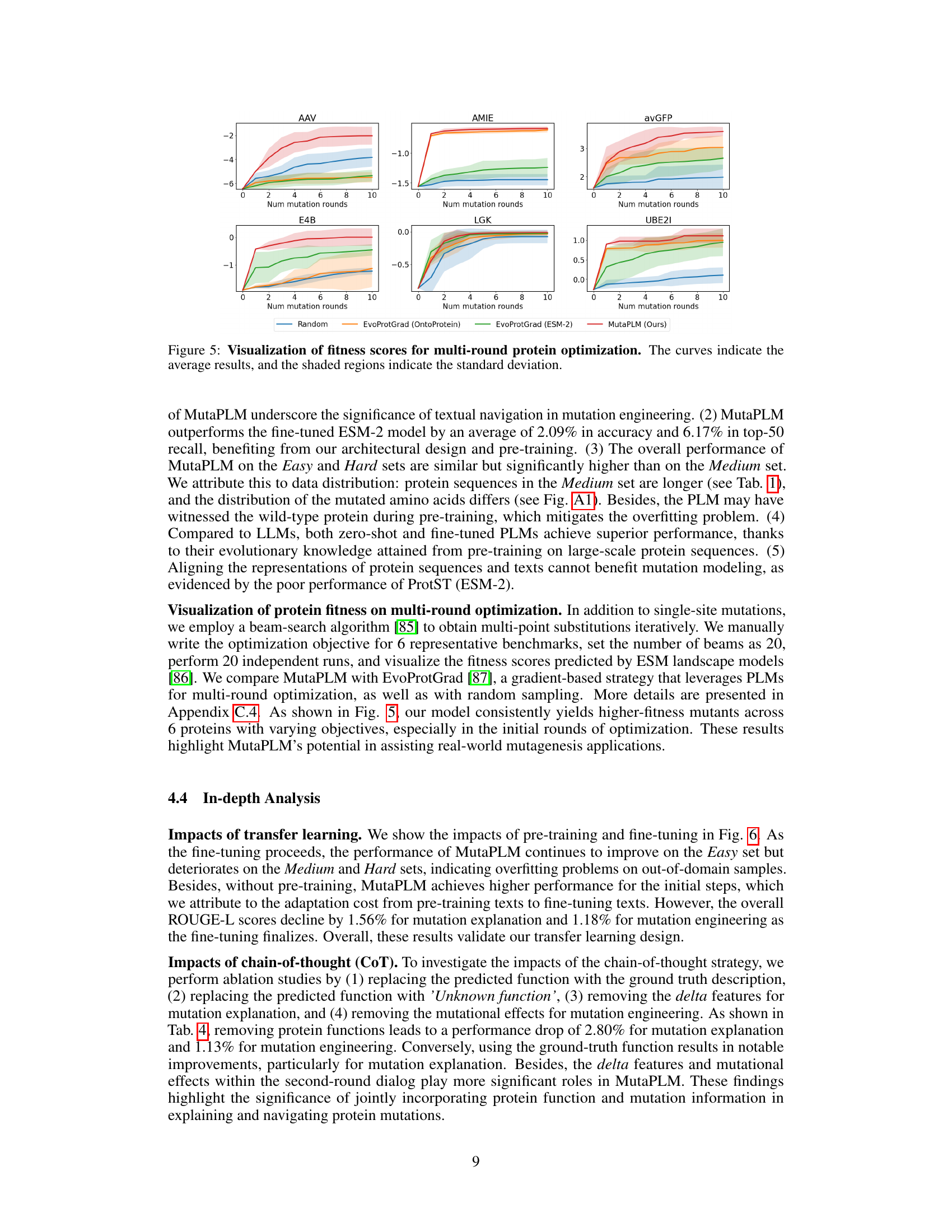
This figure visualizes the results of multi-round protein optimization using different methods. The x-axis represents the number of mutation rounds, and the y-axis represents the fitness score. Multiple lines represent different methods: Random, EvoProtGrad (OntoProtein), EvoProtGrad (ESM-2), and MutaPLM (Ours). Shaded areas show standard deviation. The figure demonstrates MutaPLM’s superior performance in achieving higher fitness scores across multiple rounds and different proteins.
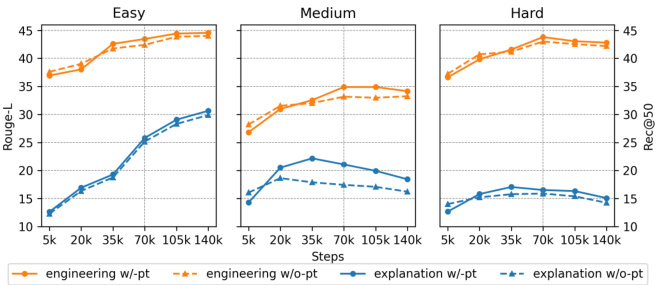
This figure displays the results of experiments evaluating the effects of pre-training and fine-tuning on MutaPLM’s performance in mutation explanation and engineering tasks. The x-axis represents the number of training steps, while the y-axis shows the ROUGE-L score (for explanation) and Recall@50 (for engineering). Separate lines are shown for models with and without pre-training. The figure shows that pre-training improves performance, particularly in the later stages of training, but also that fine-tuning can lead to overfitting if not carefully managed.
This figure shows the architecture of MutaPLM, a framework for interpreting and navigating protein mutations. It consists of two main branches: an encoding branch and a decoding branch. The encoding branch uses a protein delta network to capture explicit protein mutation representations, taking the difference between mutant and wild-type protein representations as input. The decoding branch reconstructs mutant features from the encoded differences and predicts the position and amino acid of the mutation.
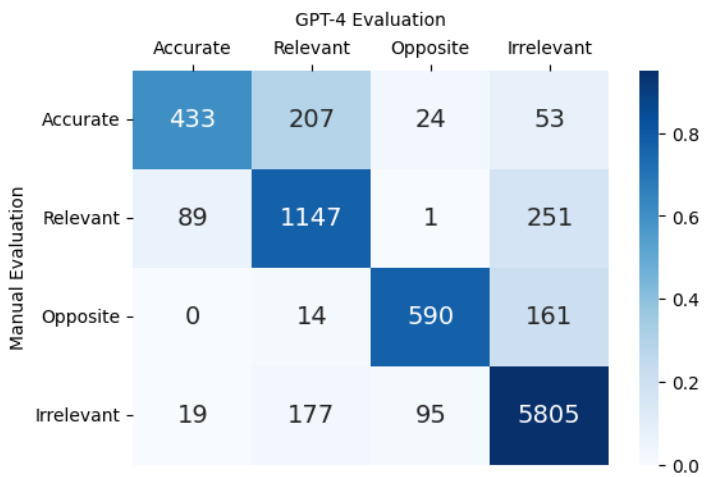
This figure presents a human-AI collaborative evaluation of MutaPLM’s mutation explanation capabilities on the MutaDescribe dataset. It displays a heatmap showing the counts of predictions categorized by a human expert as Accurate, Relevant, Opposite, and Irrelevant. The results are broken down across three difficulty levels (Easy, Medium, Hard) of the test set, demonstrating the model’s performance across different complexities of mutations.
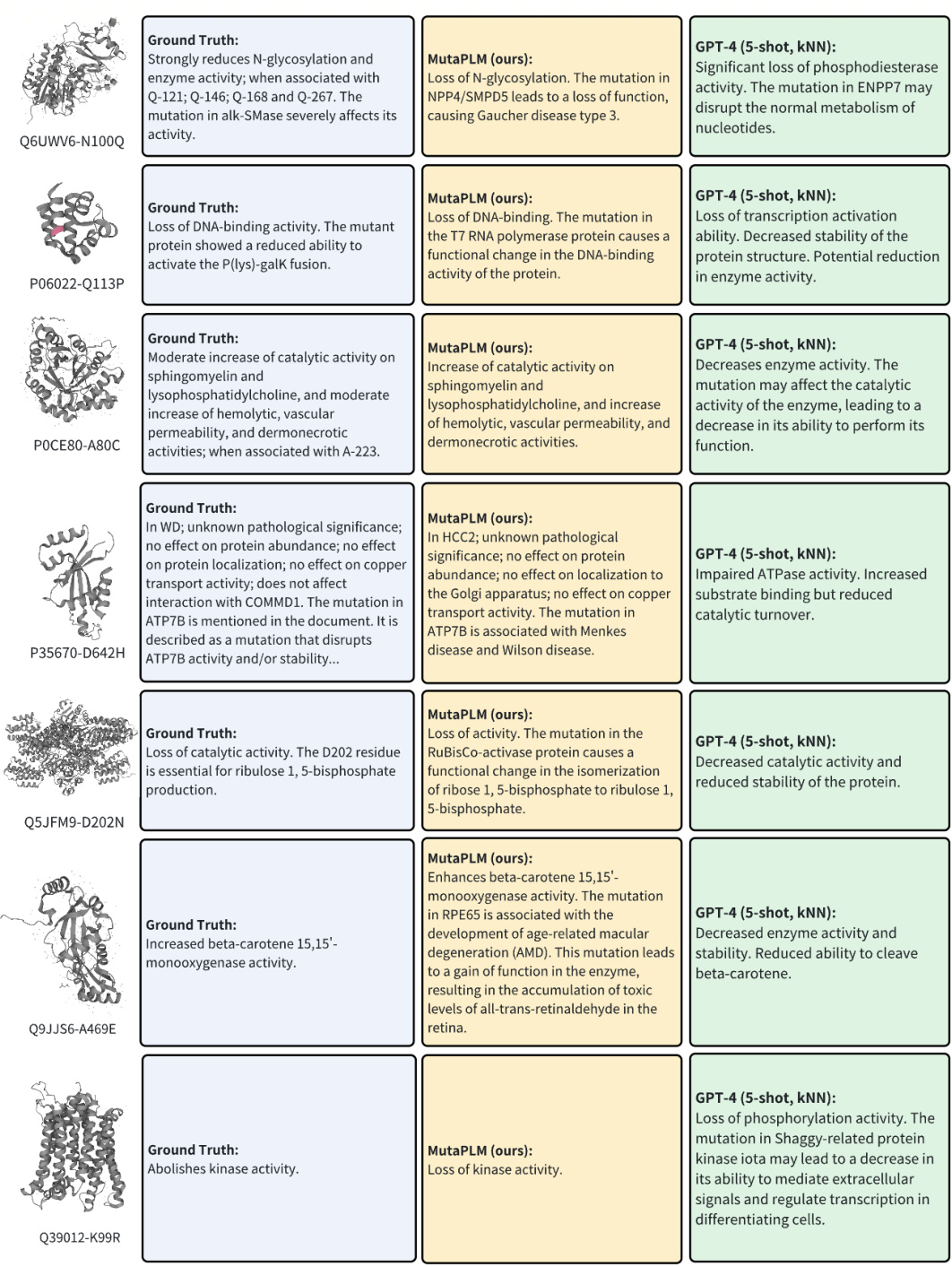
This figure illustrates the architecture of MutaPLM, a framework for interpreting and navigating protein mutations using protein language models. Panel (a) shows the encoding branch of the protein delta network, which takes the difference between the mutant and wild-type protein representations from a pre-trained PLM (protein language model) and generates a ‘delta feature’ (z∆) representing the mutation. Panel (b) depicts the decoding branch, where the delta feature is used by a delta decoder to reconstruct the mutant protein features. This information is then processed by two prediction heads that determine the mutation’s amino acid and its location in the protein sequence. In essence, the model uses the difference in representation between the wild type and the mutant as input to predict the mutation.
More on tables
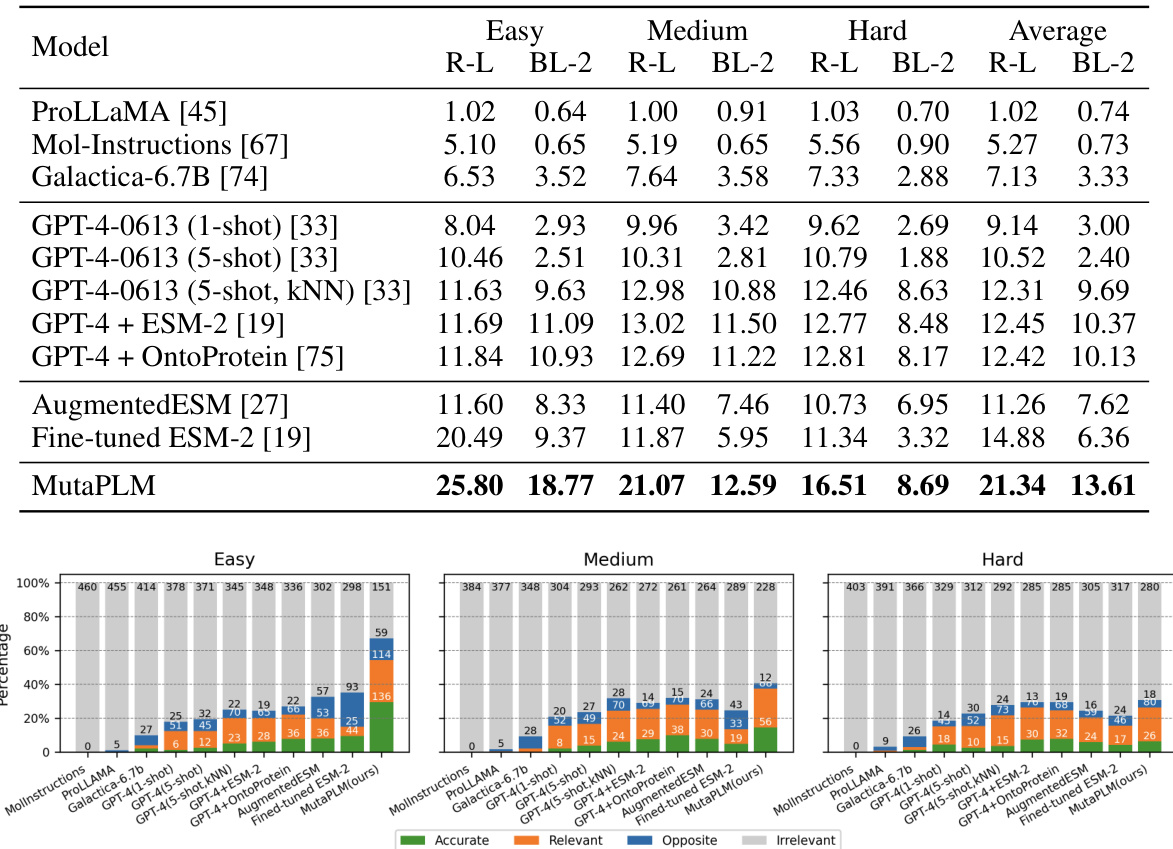
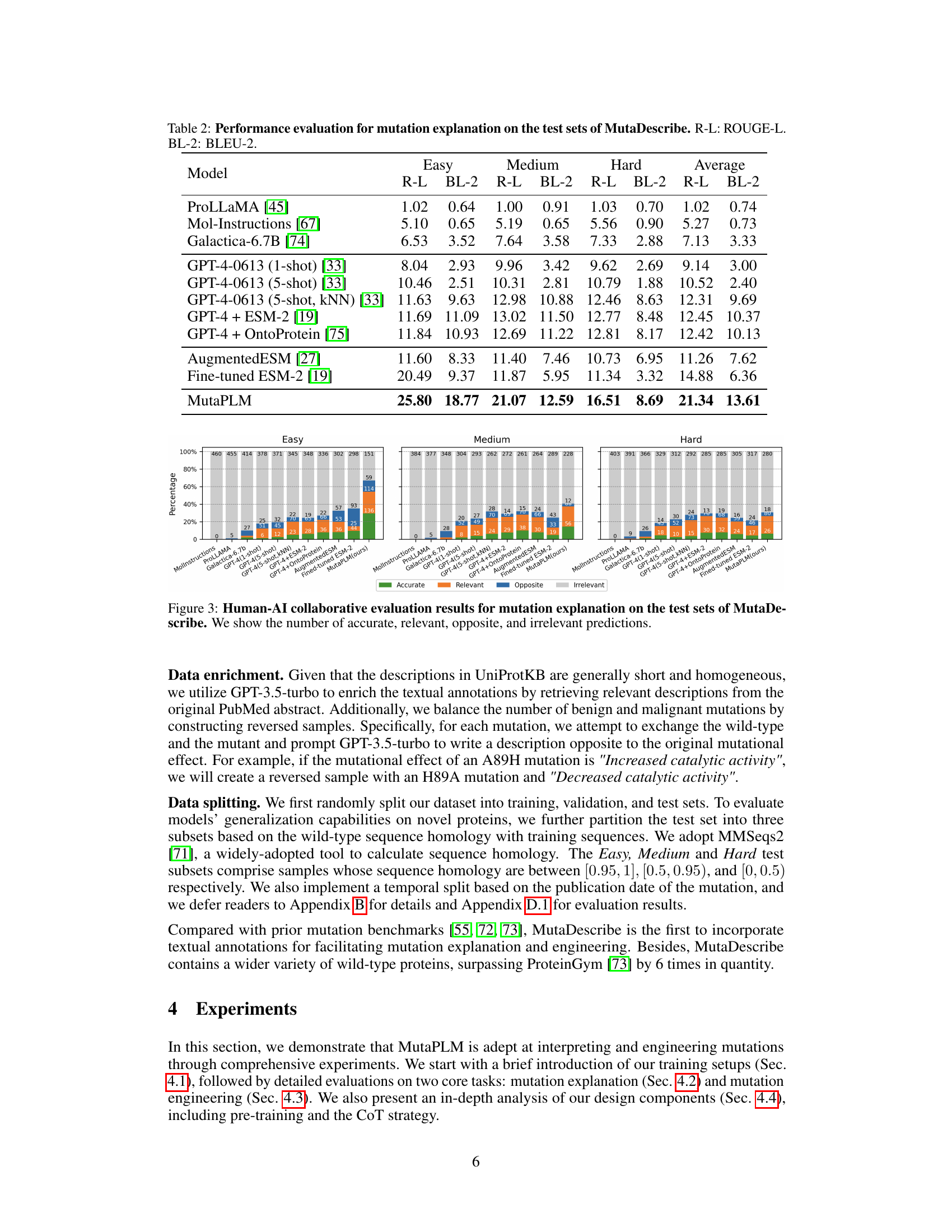
This table presents the performance evaluation results for the mutation explanation task on the MutaDescribe dataset. It compares various models (including the proposed MutaPLM) using two metrics: ROUGE-L (ROUGE-Longest Common Subsequence), a recall-oriented metric evaluating the similarity between generated and reference texts; and BLEU-2 (Bilingual Evaluation Understudy), a precision-oriented metric measuring the overlap of n-grams (in this case, bigrams). The results are broken down by three levels of test set difficulty: Easy, Medium, and Hard, each representing different levels of sequence homology between the test set and training data. The ‘Average’ column shows the performance across all three difficulty levels.
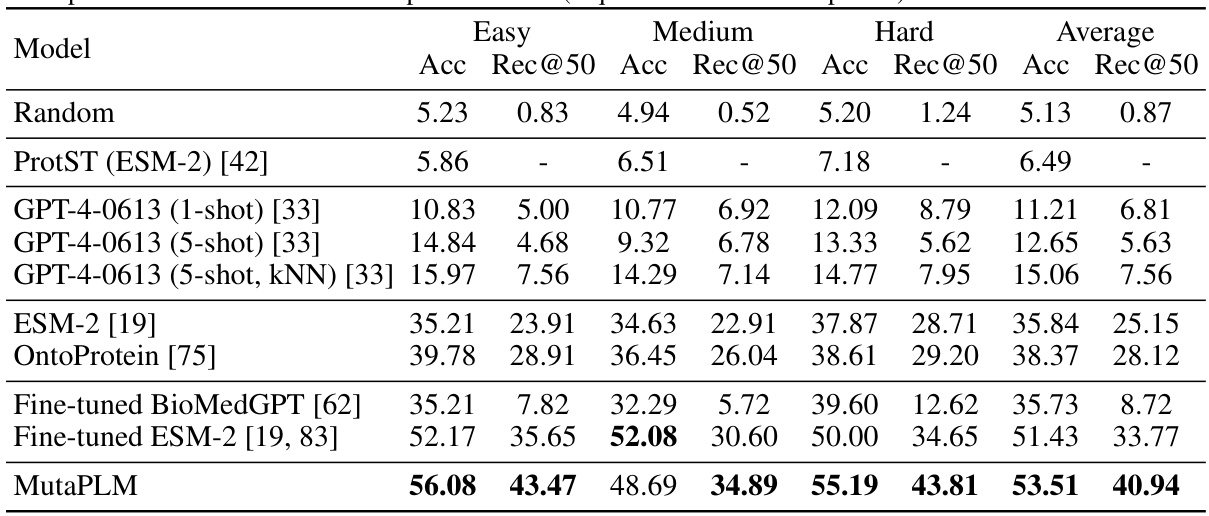
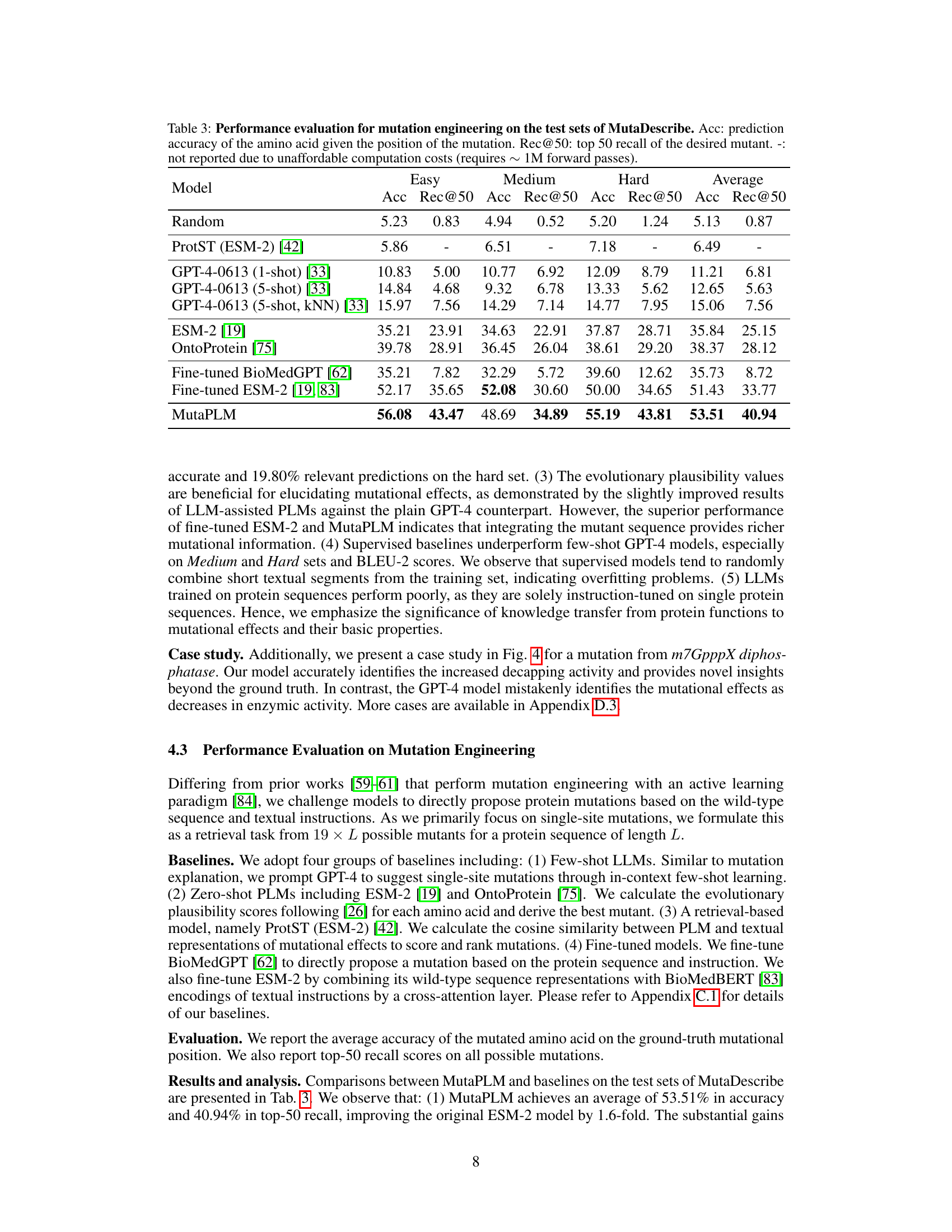
This table presents the results of mutation engineering experiments on the MutaDescribe dataset. It compares several models’ performance on two metrics: prediction accuracy (Acc) of the amino acid at the mutated position and top-50 recall (Rec@50) of the desired mutant. The models are evaluated on three difficulty levels (Easy, Medium, Hard) and their average performance across all levels is also shown. Note that some results are missing due to high computational costs.
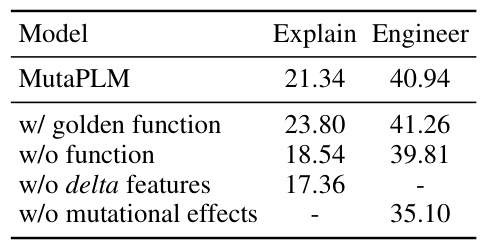
This table presents the results of ablation studies performed to evaluate the impact of different components of the MutaPLM model on its performance in mutation explanation and engineering. It shows the average ROUGE-L score for mutation explanation and the average Recall@50 score for mutation engineering, comparing the full MutaPLM model to versions where key components (the golden function, the function prediction, delta features, and mutational effects) have been removed. The results illustrate the relative contributions of these components to the overall performance of the model.

This table shows the average l2-norm of three intermediate representations generated by MutaPLM on the MutaDescribe dataset. These representations are: 1. hwt: The representation of the wild-type protein sequence. 2. hΔ: The difference between the representations of the mutant and wild-type protein sequences (mutational features). 3. zΔ: The protein delta features derived from hΔ after processing by the delta encoder. The l2-norm provides a measure of the magnitude of each representation. The table helps to illustrate that the delta encoder effectively transforms the mutational features into a more manageable and informative representation.
This table presents the performance of various models on the mutation explanation task, specifically evaluating their performance on three different test sets of the MutaDescribe dataset (Easy, Medium, and Hard). The evaluation metrics used are ROUGE-L (ROUGE-L) and BLEU-2 (BLEU-2), which are commonly used for assessing the quality of text generation. The ‘Average’ column provides an overall performance metric across all test sets.
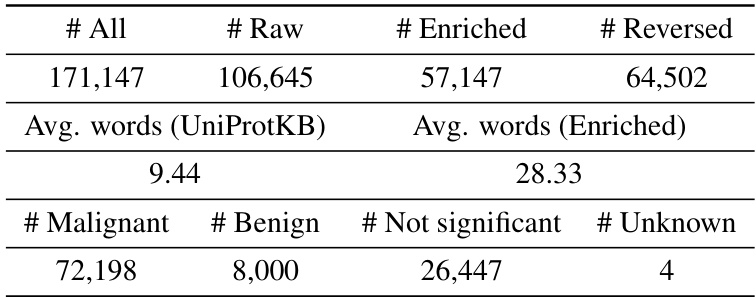
This table presents the statistical properties of the MutaDescribe dataset. It shows the total number of proteins and samples included, the average length of protein sequences in the dataset, and the average number of words used to describe the mutational effects for each mutation. This provides a quantitative overview of the dataset’s size and the descriptive detail of its annotations.
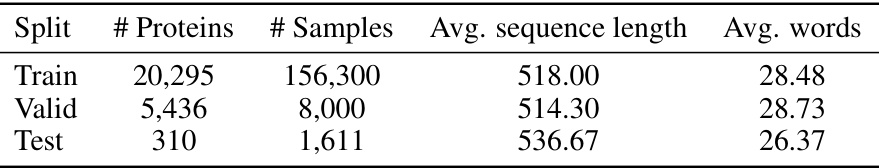
This table presents a statistical overview of the MutaDescribe dataset after it was split temporally into training, validation, and test sets. The split is based on publication date of the protein mutation data. The table shows the number of proteins and samples in each set, the average length of the protein sequences, and the average number of words used to describe the mutational effects. This information helps assess the size and characteristics of the dataset used for training and evaluation.

This table presents the performance evaluation results for the mutation explanation task on the MutaDescribe dataset’s test sets. It compares several models, including the proposed MutaPLM, using ROUGE-L (ROUGE Longest Common Subsequence) and BLEU-2 (Bilingual Evaluation Understudy) scores. The results are broken down by the difficulty level of the test set (Easy, Medium, Hard), providing a comprehensive assessment of each model’s ability to generate human-understandable explanations of mutational effects.
This table presents the performance evaluation results for the mutation explanation task on the test sets of the MutaDescribe dataset. It compares the performance of MutaPLM against various baseline models across three different difficulty levels (Easy, Medium, Hard) of the test sets. The evaluation metrics used are ROUGE-L (ROUGE Longest Common Subsequence) and BLEU-2 (Bilingual Evaluation Understudy, 2-gram precision). Higher scores indicate better performance in generating human-understandable explanations for mutational effects.

This table lists the prompts used in the mutation engineering experiments. Each prompt specifies a desired outcome or effect for a specific protein, guiding the model in suggesting mutations to achieve that outcome. The prompts vary in their level of detail and specificity, reflecting the different objectives and properties being targeted in the mutagenesis process. The proteins targeted include: AAV, AMIE, avGFP, E4B, LGK, and UBE2I.
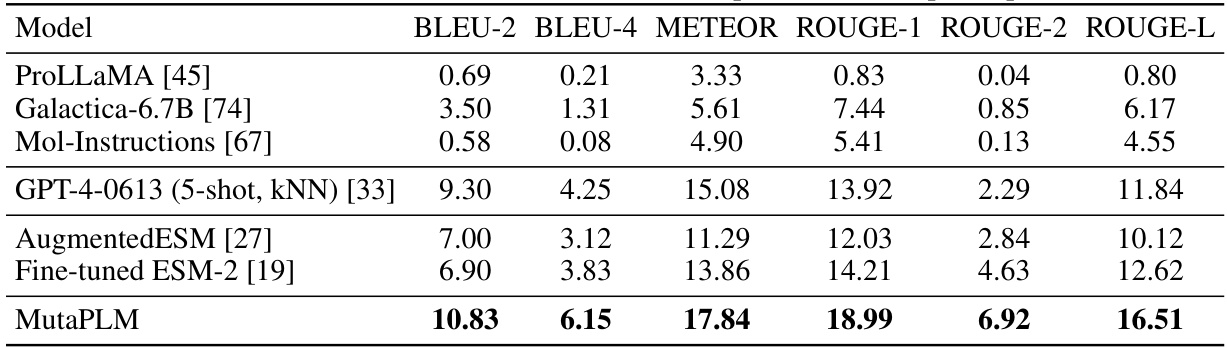
This table presents the performance of various models on the mutation explanation task using a temporal split of the MutaDescribe dataset. The models are evaluated using several metrics including BLEU-2, BLEU-4, METEOR, ROUGE-1, ROUGE-2, and ROUGE-L. A temporal split means that the data is divided based on the publication date of the mutations, allowing for evaluation of the models’ ability to generalize to mutations discovered in more recent years. The table helps to show the relative performance of each model on the task, and the superior performance of MutaPLM is highlighted.
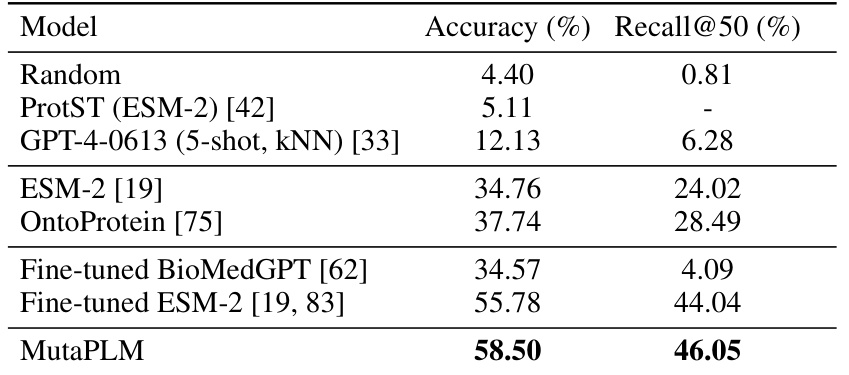
This table presents the performance of different models on the mutation engineering task using the MutaDescribe dataset. The accuracy metric reflects how often the model correctly predicts the amino acid at the mutated position. Rec@50 represents the recall at 50, indicating the proportion of times the correct mutated amino acid is found within the top 50 predictions. The ‘-’ indicates that the computation for some models was too expensive.
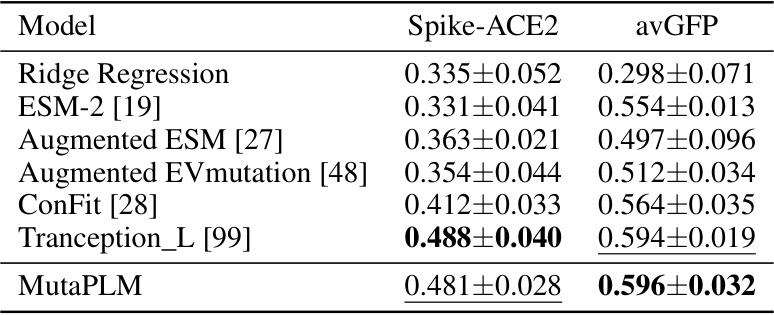
This table presents the results of experiments comparing different models’ performance on protein fitness regression tasks, specifically on two benchmarks: Spike-ACE2 and avGFP. The models tested include Ridge Regression, ESM-2, Augmented ESM, Augmented EVmutation, ConFit, Tranception_L, and MutaPLM. The performance metric used is the Spearman correlation coefficient, calculated across five runs with different random seeds. The best performing model for each benchmark is highlighted in bold.
Full paper#